Лечение прогрессирующая мышечная дистрофия дюшенна
Мышечная дистрофия Дюшенна: причины, диагностика, лечение
Этиология и встречаемость мышечной дистрофии Дюшенна. Мышечная дистрофия Дюшенна (MIM №310200) — панэтническая Х-сцепленная прогрессирующая миопатия, вызванная мутациями в гене DMD. Встречаемость составляет приблизительно 1 на 3500 новорожденных мальчиков.
Патогенез мышечной дистрофии Дюшенна. Ген DMD кодирует дистрофии, внутриклеточный белок, экспрессирующийся преимущественно в гладких, скелетных и сердечной мышце, а также в некоторых нейронах мозга. В скелетной мускулатуре дистрофии составляет часть большого комплекса связанных с сарколеммой белков, обеспечивающих устойчивость сарколеммы.
Мутации в гене DMD, вызывающие миодистрофию Дюшенна, включают крупные делеции (60-65%), крупные дупликации (5-10%) и небольшие делеции, инсерции или замены нуклеотидов (25-30%). Самые крупные делеции происходят в одной из двух горячих точек. Нуклеотидные замены встречаются по всему гену, преимущественно в динуклеотидах CpG.
De novo мутации возникают со сравнимой частотой в ходе овогенеза и сперматогенеза; наиболее крупные делеции de novo возникают в овогенезе, тогда как большинство de novo нуклеотидных замен возникает в ходе сперматогенеза.
Мутации, вызывающие фенотипическое отсутствие дистрофина, приводят к более тяжелому поражению мышц, чем мутантные аллели DMD, экспрессирующие частично функциональный дистрофии. Корреляции между генотипом и фенотипом для интеллектуального снижения не обнаружено.
Фенотип и развитие мышечной дистрофии Дюшенна
Мужчины с мышечной дистрофией Дюшенна. Миодистрофия Дюшенна — прогрессирующая миопатия, приводящая к дегенерации и слабости мышц. Начинаясь с мышц тазобедренного пояса и сгибателей шеи, мышечная слабость прогрессивно захватывает плечевой пояс и дистальные мышцы конечностей и туловища. Хотя изредка и выявляют случайно больных в период новорожденности за счет гипотонии или задержки развития, обычно больных мальчиков диагностируют в возрасте от 3 до 5 лет при появлении аномалий походки.
К 5 годам большинство пораженных детей используют приемы Говерса и имеют псевдогипертрофию мышц голеней, т.е. увеличение голеней вследствие замены мышц жировой и соединительной тканью. К возрасту 12 лет основная часть больных обездвижены в инвалидном кресле и имеют контрактуры и сколиоз. Большинство пациентов умирают от нарушения легочной функции и пневмонии; средний возраст смерти — 18 лет.
Почти 95% больных миодистрофией Дюшенна имеют те или иные кардиологические отклонения (дилатационная кардиомиопатия или электрокардиографические аномалии), а 84% имеют видимые поражения мышцы сердца при вскрытии. Хронические нарушения сердца бывают почти у 50% пациентов, изредка сердечная недостаточность вызывает у них жалобы. Хотя дистрофии также присутствует в гладких мышцах, гладкомышечные осложнения встречаются редко и включают расширение желудка, заворот кишок и гипотонию мочевого пузыря.
Больные миодистрофией Дюшенна имеют IQ примерно на 1 среднеквадратичное отклонение ниже обычного, и почти треть имеет ту или иную степень умственной отсталости. Причины этого не установлены.

Женщины с мышечной дистрофией Дюшенна
Возраст начала и тяжесть миодистрофии Дюшенна у женщин зависят от степени смещения инактивации Х-хромосомы. Если Х-хромосома, несущая мутантный аллель DMD, активна в большинстве клеток, у женщины развиваются признаки миодистрофии Дюшенна; если преимущественно активна Х-хромосома, несущая нормальный аллель DMD, женщины имеют только несколько или не имеют вообще симптомов данного заболевания.
Независимо от того, есть ли у них клинические симптомы скелетной мышечной слабости, женщины-носительницы имеют отклонения в функции сердечной мышцы, например дилатационную кардиомиопатию, дилатацию левого желудочка и электрокардиографические изменения.
Особенности фенотипических проявлений дистрофии Дюшенна:
• Возраст начала: детство
• Слабость мышц
• Гипертрофия голеней
• Небольшая интеллектуальная недостаточность
• Высокий уровень креатинкиназы сыворотки
Лечение мышечной дистрофии Дюшенна
Диагноз мышечной дистрофии Дюшенна основывается на семейном анамнезе и ДНК-анализе или мышечной биопсии с иммуногистохимическим определением дистрофина.
В настоящее время излечение мышечной дистрофии Дюшенна невозможно, хотя улучшившееся симптоматическое лечение повысило средний срок жизни от конца детства до ранней зрелости. Цели терапии — замедлить развитие болезни, обеспечить мобильность, предотвратить или исправить контрактуры и сколиоз, контролировать массу тела и улучшить функции легких и сердца.
Глюкокортикоидная терапия может замедлять развитие заболевания в течение нескольких лет. Исследуют несколько видов экспериментального лечения, включая генную передачу. Большинство больных нуждается также в расширенном консультировании, так как имеют дело с психологическими эффектами хронической фатальной болезни.
Риск наследования мышечной дистрофии Дюшенна
Третья часть матерей, родивших единственного больного сына, сами являются носительницами мутаций в гене DMD. Тем не менее определение носительства остается трудной задачей, поскольку в настоящее время доступные молекулярные методы не обнаруживают небольшие мутации типа однонуклеотидных замен. Определение риска носительства в семьях без найденной делеции или дупликации основывается на анализе сцепления, серии анализов уровня креатинкиказы сыворотки крови и мозаичной экспрессии дистрофина в образцах биопсии мышц (из-за случайной инактивации Х-хромосомы). При консультировании в связи с оценкой риска повторения нужно принимать во внимание высокую частоту мозаицизма в половых клетках (приблизительно 14%).
Если мать — носитель, каждый сын имеет 50% риск развития мышечной дистрофии Дюшенна, а каждая дочь — 50% риск унаследовать мутацию DMD. Отражая случайную природу инактивации Х-хромосомы, дочери, унаследовавшие мутацию в гене DMD, имеют низкий риск мышечной дистрофии Дюшенна; тем не менее по причинам, полностью непонятным, риск сердечных аномалий может достигать 50-60%. Если мать не носительница по результатам ДНК-тестирования, у нее остается приблизительно 7% риска родить мальчика с мышечной дистрофией Дюшенна вследствие полового мозаицизма. Для этих матерей показаны генетическое консультирование и, возможно, пренатальная диагностика.
Пример мышечной дистрофии Дюшенна. А.И., 7-летний мальчик, проходит обследование в связи с легкой задержкой развития. У него затруднения при подъеме по ступенькам, беге, снижение силы и выносливости при интенсивной физической нагрузке. Его родители, два брата и сестра полностью здоровы; другие члены семьи аналогичных жалоб не имеют. При осмотре выявлены затруднения при подпрыгивании, приемы Говерса (последовательность движений, облегчающих вставание с пола), слабость проксимальных мышц, переваливающаяся («утиная») походка, уплотнение ахилловых сухожилий и заметно гипертрофированные мышцы голеней. Уровень креатинкиназы сыворотки крови оказался в 50 раз выше нормы.
Поскольку анамнез и данные медицинского осмотра, включая повышенный уровень креатинкиназы, позволили предположить миопатию, ребенок для дальнейшего обследования направлен в клинику нейрогенетики. Результаты биопсии мышц показали выраженное изменение размера мышечных волокон, некроз волокон, разрастание жировой и соединительной ткани и отсутствие окрашивания на дистрофии. На основании этих результатов ребенку установлен предварительный диагноз мышечной дистрофии Дюшенна, и он тестирован на делеции в гене дистрофина; оказалось, что у него имеется делеция с 45 по 48 экзон.
Дальнейшее обследование показало, что его мать была носителем этой делеции. Соответственно семье дано заключение, что риск для сыновей составляет 50%, риск для дочерей низкий, но зависит от смещения Х-инактивации, а риск носительства для дочерей равен 50%. Поскольку статус носительницы у матери дает повышенный риск сердечных осложнений, она направлена на кардиологическое обследование.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
- Мышечная дистрофия Дюшенна: причины, диагностика, лечение
- Семейный полипоз кишечника: причины, диагностика, лечение
- Семейная гиперхолестеринемия: причины, диагностика, лечение
- Синдром ломкой Х-хромосомы: причины, диагностика, лечение
- Недостаточность глюкозо-6-фосфат дегидрогеназы (Г6ФД): причины, диагностика, лечение
- Наследственный гемохроматоз: причины, диагностика, лечение
- Гемофилия: причины, диагностика, лечение
- Наследственный неполипозный рак кишечника (ННРК): причины, диагностика, лечение
- Болезнь Гиршспрунга: причины, диагностика, лечение
- Голопрозэнцефалия: причины, диагностика, лечение
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Прогрессирующая мышечная дистрофия Дюшенна

Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
- Причины
- Симптомы
- Осложнения
- Диагностика
- Лечение мышечной дистрофии Дюшенна
- Стандартная терапия
- Экспериментальная терапия
Общие сведения
Прогрессирующая мышечная дистрофия Дюшенна — тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Прогрессирующая мышечная дистрофия Дюшенна
Причины
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
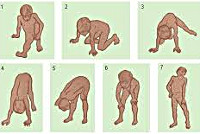
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Осложнения
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК:
- ЭФИ. Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы.
- Генетическая диагностика. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
- Биопсия. В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
- Другие исследования. Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Лечение мышечной дистрофии Дюшенна
Стандартная терапия
Терапия, применяемая в клинической практике, включает симптоматическое и патогенетическое направление. В рамках данных направлений применяется медикаментозная терапия, физическая реабилитация, респираторная поддержка:
- Кортикостероиды. Основная роль в лечении мышечной дистрофии Дюшенна на сегодняшний день отводится глюкокортикостероидам, которые назначаются как способным, так и не способным к самостоятельному передвижению пациентам. ГКС помогают замедлить прогрессирование мышечной слабости, оказывают умеренный пульмопротективный и кардиопротективный эффект, снижают риск развития ортопедических осложнений. Из-а большого количества побочных эффектов глюкокортикостероидной терапии необходим тщательный мониторинг состояния ребенка, своевременная коррекция дозы и схемы приема препарата.
- Метаболическая терапия. Направлена на улучшение обменных процессов в скелетной мускулатуре, костях, сердечной мышце, печени, снижение побочных эффектов от приема ГКС. Включает назначение витаминов группы В, левокарнитина, препаратов Са, витамина D.
- Физическая терапия. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия, пассивная и активная растяжка. Рекомендуется использование ортезов, вертикализатора, специальных шин, занятия лечебным плаванием.
- Респираторная поддержка. Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Экспериментальная терапия
Перспективным направлением является разработка и использование препаратов, нацеленных на восстановление синтеза или замещение дистрофина. В 2020 г. в России был зарегистрирован первый таргетный препарат для лечения мышечной дистрофии Дюшенна — аталурен, который подходит 10% пациентам с нонсенс-мутацией (стоп-кодоном). Препарат позволяет возобновить синтез белка дистрофина в организме, требует пожизненного приема. Другие аналогичные лекарственные средства (этеплирсен, голодирсен) в РФ пока не одобрены к применению. Положительные результаты на стадии клинических испытаний у пациентов с миодистрофией Дюшенна показал микродистрофин. Идет поиск и апробация методов заместительной генной терапии.
Прогноз и профилактика
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Миодистрофия Дюшенна: можно ли противостоять генетике?

Генетические заболевания, сцепленные с полом — это специфическая группа патологий, развивающихся в результате мутаций в половых (непарных) хромосомах, и передаваемых по наследству от родителей к детям. Примером служит миодистрофия Дюшенна — патология, связанная с материнской Х-хромосомой в генотипе мальчиков и проявляющаяся практически всегда только у них.
Мутация связана с повреждением DMD-гена, кодирующего синтез специфического мышечного белка — дистрофина. Встречается она нечасто, примерно 1 случай на 4-5 тысяч новорожденных мальчиков по всему миру. Но последствия мутации очень серьезны, приводят к ранней инвалидности, прогрессирующим костно-мышечным нарушениям, приводящим в итоге к гибели в молодом возрасте, если ребёнком активно не заниматься.

Какие проблемы с кожей помогут решить средства с гиалуроновой кислотой?
Сухость кожи, чувство стянутости — всё это и другие проблемы, которые решают средства с гиалуроновой кислотой.
В чем суть патологии «миодистрофия Дюшенна»

В связи с мутацией гена, кодирующего мышечный белок дистрофин, формируется аномалия мышечного тонуса и силы. Другими словами, для патологии типично развитие прогрессирующей слабости мышц, впервые возникающей в относительно раннем возрасте, примерно после трех лет. По мере прогрессирования болезни ребёнок утрачивает возможности двигаться самостоятельно, у него пропадают навыки самообслуживания, если не проводить активные лечебные и реабилитационные мероприятия. Опасность этой патологии еще и в том, что патология постепенно вовлекает в процесс группу дыхательных мышц, что грозит смертью от паралича дыхания в раннем возрасте, обычно после 20-25 лет.
Ближе к подростковому периоду патология начинает активнее прогрессировать, и после 10-15 лет дети уже не могут передвигаться сами, без инвалидных кресел. Сегодня пока нет эффективных методов лечения данной патологии, возможно только поддерживающее лечение, которое позволяет многим больным доживать до 30 лет и более. Но в нашей стране пока только разрабатываются и внедряются новые программы для таких детей. Однако перспективы лечения есть, многие лаборатории мира занимаются разработкой новых препаратов.
Если в семье родился такой ребёнок.
Проблема данной болезни в том, что диагностика с раннего возраста крайне затруднена, и первые признаки выявляются уже ближе к 3-м годам, когда мальчик начинает отставать в развитии — как речевом, так и физическом, — возникают определенные трудности с двигательной функцией. Но важно, чтобы родители приняли диагноз и продолжили жить дальше, активно занимались ребёнком и жили привычной жизнью, не ограничивая сына в каких-либо сферах. Ребёнок должен посещать садик и школу, общаться с друзьями, и относиться к нему нужно соответственно состоянию на каждый момент времени.
Сегодня в нашей стране появился проект в поддержку этой болезни, созданный семьей, в которой есть такой ребёнок — “Red Balloons”. Он объединяет семьи с такими детьми и подростками, позволяет им обмениваться информацией, подыскивать врачей и специалистов по реабилитации, получать помощь и психологическую поддержку. Участники Red Balloons помогают пошаговыми инструкциями тем семьям, которые столкнулись с подобным диагнозом и не знают, как быть дальше.
Проблемы лечения

Для нашей страны это заболевание еще относительно редкое, и поэтому нет современных специализированных центров, занимающихся прицельно именно миодистрофией Дюшенна. Проблема заключается и в поисках специалистов — генетиков, неврологов и реабилитологов, особенно в регионах, которые могли бы оказать помощь, быстро поставить диагноз и разработать индивидуальную программу лечения с реабилитацией.
В интернете данные о патологии разрозненные и отрывочные, в связи с чем проект Red Balloons занимается еще и информационным обеспечением родителей полноценной и подробной информацией о болезни. На примере одной семьи родители, воспитывающие сына с этим редким заболеванием, дают другим родителям понять, что такое миодистрофия Дюшенна, как ее заподозрить, что делать дальше и как заниматься реабилитацией.
В Европе и США родительские сообщества более развиты, много и научных институтов, занимающихся разработкой методик лечения, фармакологические компании активно разрабатывают препараты против болезни, а также средства поддерживающей терапии. В нашей стране большинство специалистов, занимающихся данной патологией, работает в столице, в регионах врачей, хорошо разбирающихся в болезни, — единицы. У большинства семей после консультации у генетика и постановки диагноза лечением занимается невролог, и зачастую применяются совсем не те методы и препараты, которые необходимы.
А именно постоянное лечение и реабилитация необходимы таким детям и подросткам, особенно когда болезнь прогрессирует. Есть часть процедур и манипуляций, которые таким детям противопоказаны, например, инъекции внутримышечно или некоторые виды массажа. Поэтому важно проконсультироваться с опытным врачом, и не предпринимать до этого попыток самолечения.
Проблемы детей и родителей
При наличии подобного диагноза крайне важна помощь государства и полноценное лечение, но получить поддержку можно только после выставления инвалидности, а на прохождение комиссий порой уходит много драгоценного времени. В случае установления инвалидности вполне можно получить индивидуальные программы по реабилитации мальчиков — ортопедическую обувь, специальные коляски, особенно, если нет возможности покупать их самим.
Наиболее важным этапом является поддерживающее лечение — оно складывается из медикаментозной поддержки и физиотерапии, и это самый сложный из этапов лечения. Покупать препараты нередко нужно самим, нет грамотных специалистов в регионах, нет специальных приборов и устройств, которые помогут в нормализации двигательной функции. У трети таких детей имеются еще и речевые проблемы, устранять которые должны не логопеды, а профессиональные дефектологи. Это один из важных этапов в социализации ребёнка.
Лечение и его трудности

Самой сложной сегодня проблемой является лекарственное обеспечение, так как не все препараты, применяемые в терапии, разрешены к применению в нашей стране. В России применяют в лечении преднизолон, который устарел и имеет массу побочных эффектов, а более современный и эффективный препарат — Дефлазакорт — родителям необходимо своими силами доставлять из стран Европы. Там он активно применяется и относится к золотому стандарту терапии этой патологии. Он помогает в поддержании силы мышц. Родители таких малышей бьются над тем, чтобы был создан единый реестр детей с подобными патологиями, что позволило бы фармкомпаниям приходить на рынок с препаратами для лечения этой категории пациентов.
Сегодня разрабатываются различные препараты против миодистрофии Дюшенна, но они эффективны далеко не у всех пациентов. Есть определенные продвижения в разработке препаратов, так в США одобрен к применению Этеплирсен, а в странах Европы — Аталурен. У нас в стране они не применяются из-за того, что не включены в реестр лекарственных препаратов.
Не так давно были обнародованы данных исследований новых препаратов, часть из них тестируется уже в клиниках, а для других пока проводятся лабораторные испытания. Основа препаратов — это часть поврежденного гена, который при введении в организм должен частично заменить в работе поврежденные участки в собственных клетках. По мнению создателей, такие препараты могут замедлить прогрессирование дистрофии мышц.
Источник https://meduniver.com/Medical/genetika/distrofia_dushenna.html
Источник https://www.krasotaimedicina.ru/diseases/zabolevanija_neurology/Duchenne-muscular-dystrophy
Источник https://medaboutme.ru/articles/miodistrofiya_dyushenna_mozhno_li_protivostoyat_genetike/