Редкие нарушения свертывания крови: наследственный дефицит факторов свертывания крови II, VII, X
К редким нарушениям свертывания крови (РНСК) относят моногенные коагулопатии, вызванные дефицитом плазменных белков, участвующих в гемостазе, не относящиеся к болезни Виллебранда и гемофилии А или В. РНСК включают наследственные дефициты или аномалии фибриногена, протромбина (фактора II), факторов свертывания крови V, VII, X, XI, XII, XIII. Все эти нарушения в подавляющем большинстве случаев приводят к нарушениям формирования фибрина.
Причинами развития РНСК является, как правило, рецессивное наследование уникального нарушения или редких нуклеотидных изменений в генах, кодирующих коагуляционные факторы, или в белках, необходимых для посттрансляционных модификаций данных факторов. РНСК наиболее распространены в этнических группах, в которых приняты близкородственные браки, вследствие большей вероятности гомозиготного носительства дефекта гена.
Опубликованные описания РНСК до недавнего времени исторически состояли из исследований случай-контроль или малочисленных когортных исследований. Однако, в течение последних 10 лет появилось несколько специфических регистров (European Network of Rare Bleeding Disorders, Peyvandi et al, 2012; the North American Rare Bleeding Registry, Acharya et al, 2004; RCD-Registries: Herrmann et al, 2006, 2009; Ivaskevicius et al, 2007; Bernardi et al, 2009), позволивших улучшить понимание РНСК. Создание международной базы РНСК способствовало определению четких лабораторных критериев тяжести большинства РНСК, которые были разработаны под эгидой Международного Общества по Тромбозу и Гемостазу (ISTH) в 2012 году.
Наследственный дефицит фактора свертывания крови II (гипопротромбинемия) — геморрагическое заболевание, характеризующееся снижением активности протромбина в плазме, возникающим вследствие генетических дефектов, обуславливающих количественные (гипопротромбинемия) или качественные (диспротромбинемия) нарушения фактора свертывания крови II (FII). Наследуется по аутосомно-рецессивному типу. Распространенность в большинстве стран составляет 1:2 000 000.
FII — это гликопротеид, который образуется в печени в присутствии витамина K. Под влиянием активированного фактора свертывания X (FXа) в инициирующей фазе коагуляционного каскада и протромбиназного комплекса (фаза амплификации) протромбин превращается в тромбин. Тромбин, в свою очередь, активирует другие плазменные факторы свертывания и тромбоциты с конечным формированием фибринового сгустка. Кроме того, тромбин участвует в активации ингибиторов свертывания и в регуляции фибринолиза.
Дефицит FII обусловлен мутациями гена F2, кодирующего протромбин. Не существует прямой корреляции между генотипом F2 и фенотипом заболевания. Гемостатически достаточный уровень FII составляет около 40%. Период полувыведения FII — около 60 часов.
Наследственный дефицит фактора свертывания крови VII (гипопроконвертинемия) — геморрагическое заболевание, возникающее вследствие генетически обусловленного снижения активности фактора свертывания крови VII (FVII) в плазме. Наследуется по аутосомно-рецессивному типу. Распространенность составляет 1:300 000—1:500 000.
Физиологическая роль FVII заключается в инициации процесса свертывания в зоне повреждения сосудистой стенки. В комплексе с тканевым фактором активированный FVII активирует FX и FIX, участвующие в генерации тромбина. Период полувыведения FVII составляет 4—6 часов, достаточный гемостатический уровень — не менее 10%. При тяжелых травмах клинически значимое кровотечение может развиться при уровне FVII более 20%.
Наследственный дефицит фактора свертывания крови Х — геморрагическое заболевание, возникающее вследствие генетически обусловленного количественного или качественного дефекта FX, приводящего к снижению активности FХ в плазме. Наследуется по аутосомно-рецессивному типу. Средняя распространенность заболевания составляет 1:1 000 000 [1].
Активация FX происходит в инициирующей фазе свертывания крови при участии комплекса тканевой фактор — активированный FVII и в фазе амплификации c теназным комплексом. Активированный FX (FXa) и его кофактор, фактор свертывания V, входят в состав протромбиназного комплекса, который активирует протромбин. Период полувыведения FX составляет 30—50 часов, достаточный гемостатический уровень: кровотечения не наблюдаются у лиц с активностью FХ > 40% [1].
Облачная МИС «МедЭлемент»

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран
— 800 RUB / 5500 KZT / 27 BYN — 1 рабочее место в месяц
Облачная МИС «МедЭлемент»

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место — 800 RUB / 5500 KZT / 27 BYN в месяц
+7 938 489 4483 / +7 707 707 0716 / + 375 29 602 2356 / office@medelement.com
Мне интересно! Свяжитесь со мной
Классификация
Классификация
Существует 3 формы тяжести РНСК в зависимости от активности FII, FVII, FX, определяемой лабораторно (табл.1).

Таблица 1. Формы РНСК по степени тяжести
Тяжелый дефицит факторов, как правило, связан с развитием тяжелых спонтанных кровотечений/кровоизлияний.Умеренный дефицит факторов проявляется в виде легких/умеренных спонтанных и посттравматических эпизодов кровотечений. Легкие формы дефицита факторов в большинстве случаев протекают бессимптомно [2].
Клиническая картина
Cимптомы, течение
Клинические признаки
Типичными для РНСК являются кровотечения/кровоизлияния, возникающие спонтанно или вследствие травмы. Геморрагический синдром представлен кровотечениями из слизистых (носовые, десневые, луночковые), экхимозами, гематомами мягких тканей различной локализации, кровотечениями во время и после хирургических вмешательств, меноррагиями, гематуриями, реже – кровоизлияниями в суставы (гемартрозами). Кроме того, при значительных дефицитах факторов у пациентов отмечаются жизнеугрожающие кровотечения, к которым относятся [3]:
- кровотечения/кровоизлияния в ЦНС;
- кровотечения/кровоизлияния в ЖКТ;
- кровотечения/кровоизлияния в шею/горло;
- забрюшинная гематома
При гипопротромбинемии с активностью FII < 10%, как правило, отмечаются более тяжелые кровотечения по сравнению с кровотечениями, при активности FII ≥ 10% наиболее типичны легкие и умеренные кровотечения из слизистых оболочек. Пациенты с активностью FII менее 4% не описаны.
Для диспротромбинемии характерна слабая взаимосвязь между клиническим и лабораторным фенотипами заболевания. У гетерозиготных носителей дефицита FII в большинстве случаев определяется активность FII в пределах 40-75%, течение заболевания бессимптомное [4].
При гипопроконвертинемии приблизительно в 60% случаев течение заболевания бессимптомное, и поводом для диагностики является случайное обнаружение увеличения ПВ. Кровоизлияние в ЦНС регистрируется у 3-10% пациентов с дефицитом FVII.
Тяжелые кровотечения наиболее характерны при активности FVII ≤ 1%. У пациентов с активностью FVII > 1% отмечаются, как правило, умеренные и легкие кровотечения из слизистых оболочек или бессимптомное течение заболевания. Однако в ряде случаев регистрируются тяжелые проявления геморрагического синдрома у лиц с активностью FVII > 20%, что говорит об отсутствии прямой корреляции между клиническим и лабораторным фенотипом заболевания.
У гетерозиготных носителей дефицита FVII в большинстве случаев определяется активность FVII в пределах 40-60% с бессимптомным течением заболевания [5, 6].
При дефиците FX тяжелые кровотечения наиболее характерны для пациентов с активностью FX ≤ 10%. У пациентов с активностью FX > 10%, как правило, отмечаются умеренные и легкие кровотечения из слизистых оболочек, кровотечения после хирургических вмешательств или бессимптомное течение заболевания. Кровоизлияния в ЦНС, кровотечения из слизистых ЖКТ, гемартрозы отмечаются у пациентов с активностью FX ≤ 2%. Тем не менее, в ряде случаев регистрируются тяжелые проявления геморрагического синдрома у лиц с активностью FX 0,1 — 39%, что указывает на отсутствие прямой корреляции между клиническим и лабораторным фенотипом заболевания [7].
У гетерозиготных носителей дефицита FX в большинстве случаев активность FX составляет около 50%, и проявлений заболевания нет.
Диагностика
Диагностика
Диагностика РНСК начинается со сбора персонального и семейного анамнеза геморрагического синдрома. При сборе анамнеза заболевания рекомендуется выяснять наличие жалоб на легко появляющиеся экхимозы и гематомы в раннем детстве, спонтанные кровотечения (в том числе гематомы различной локализации, кровотечения из слизистых), длительные кровотечения после травм или хирургического вмешательства [8].
Примерно у 2/3 больных нет указаний на геморрагические проявления у близких родственников. Необходимо обращать внимание на наличие геморрагических проявлений в неонатальном периоде в виде кефалогематом, внутричерепных кровоизлияний, кровоточивости и длительном заживлении пупочной ранки; у грудных детей – экхимозов, не связанных со значимой травмой, гематом мягких тканей после незначительных ушибов или спонтанных [9]. Важно обращать внимание на несоответствие выраженности геморрагических проявлений тяжести предшествовавшей травмы, на рецидивы кровотечений после первичной остановки, не связанные с повторной травмой, массивные и/или множественные гематомы, системность геморрагических проявлений (проявления различной локализации). У лиц, с незначительными дефицитами факторов свертывания крови кровотечения могут отсутствовать до первой травмы или хирургического вмешательства. Сбор жалоб и анамнеза позволит определить объем обследования пациента.
Лабораторная диагностика
Для диагностики РНСК обязательно проведение поэтапного лабораторного коагулологического исследования. Получение максимального количества данных способствует верификации диагноза и исключению приобретенных дефицитов факторов свертывания крови II, VII, X, а также исключению дефицитов других факторов свертывания крови.
При подозрении на геморрагические состояния первым этапом лабораторной диагностики является коагулологический скрининг, при котором определяется активированное частичноео тромбопластиновое временя (АЧТВ), протромбиновое временя (ПВ), тромбиновое временя (ТВ), концентрация фибриногена (по Клауссу), временя кровотечения (ВК) стандартизованным методом (например, по Айви или с применением анализаторов PFA-100, PFA-200) и количество тромбоцитов по Фонио [10]. Показатели скрининговых тестов для дефицита FII, FVII, FX представлены в таблице 2.

Таблица 2. Скрининговые коагулологические тесты для диагностики РНСК
Если выявлено увеличение ПВ, проводится второй этап диагностики: определение активности факторов II, V, VII, IX, X, VIII, тест на волчаночный антикоагулянт, анализ функции тромбоцитов и активности фактора Виллебранда [2].
Диагноз РНСК считается установленным при выявлении изолированного снижения активности соответствующих факторов — FII, FVII или FX.
При любом удлинении ПВ необходимо исключить все виды приобретенного дефицита факторов протромбинового комплекса, в первую очередь обусловленные тяжелой патологией печени, механической желтухой, токсическим действием антикоагулянтов непрямого действия, циркулирующими антифосфолипидными антителами, эндогенным дефицитом витамина К, тяжелыми инфекциями, онкологического заболеваниями, системным амилоидозом и нефротическим синдромом [11].
Третий этап диагностики — проведение молекулярно-генетической диагностики мутаций генов FII, FVII, FX.
Рекомендуется для уточнения диагноза и проведения дифференциальной диагностики с другими заболеваниями с возможным наличием геморрагического синдрома.
Инструментальная диагностика позволяется визуализировать кровотечения/кровоизлияния различных локализаций, а также выявить осложнения, развившееся вследствие геморрагических проявлений. По показаниям проводятся следующие обследования. По показаниям проводится:
- эзофагогастродуоденоскопия,
- ультразвуковое исследование сустава,
- ультразвуковое исследование органов брюшной полости,
- ультразвуковое исследование мочевыводящих путей,
- ультразвуковое исследование органов малого таза,
- ультразвуковое исследование забрюшинного пространства,
- магнитно-резонансная томография сустава, мягких тканей,
- магнитно-резонансная томография головного мозга,
- рентгенография сустава,
- компьютерная томография органов грудной клетки,
- компьютерная томография головного мозга,
- риноскопия эндоскопическая,
- ректороманоскопия,
- кольпоскопия,
- гистероскопия,
- колоноскопия.
Для подтверждения наличия геморрагических проявлений (или их последствий) также рекомендуется проведение консультации специалистов. По показаниям возможны консультации:
- травматолога-ортопеда
- хирурга
- уролога
- невролога
- оториноларинголога
- стоматолога
У части больных могут быть выполнены интегральные тесты оценки гемостаза: тромбодинамика, тромбоэластография, тест генерации тромбина. Целесообразно выполнение интегральных тестов в случае невозможности проведения полноценного двухэтапного коагулологического исследования, а также в некоторых случаях для контроля за проводимой терапией.
Критерии диагноза РНСК, независимо от наличия геморрагического синдрома в персональном или семейном анамнезе [2, 12]:
- отсутствие данных о наличии приобретенного дефицита факторов свертывания крови;
- изолированное снижение активности FII/FVII/FX ниже 50%;
- наличие мутаций генов FII, FVII, FX.
Диагноз наследственного РНСК устанавливается при наличии не менее двух из трех вышеперечисленных критериев.
Лечение
Лечение
Основой лечения пациентов с РНСК является специфическая заместительная терапия препаратами, содержащими дефицитный (со сниженным количеством или отсутствующий) фактор свертывания. Заместительная терапия проводится как по факту кровотечения (по требованию), так и профилактически при тяжелых формах заболевания. Расчет дозы и режимы введения препаратов представлены в таблицах 3 и 4 [12].
При наследственном дефиците FII рекомендуется проведение специфической заместительной терапии неактивированными препаратами, содержащими FII. Строго рекомендуется использование плазматических очищенных вирусинактивированных концентратов протромбинового комплекса (КПК).
КПК вводятся внутривенно. В настоящее время в РФ зарегистрированы 2 препарата КПК, содержащие FII: КПК, содержащий факторы свертывания крови II, VII, IX, X в комбинации (протромбиновый комплекс) и КПК, содержащий факторы свертывания крови II, IX и X в комбинации.Используется болюсная инфузия со скоростью, рекомендованной производителем. Активность КПК, обычно, указывается по активности содержащегося в них фактора свертывания крови IX (FIX). Поэтому, необходимо отдельно уточнять содержание FII, которое указывается в инструкции. Как правило, КПК содержат приблизительно равное количество FIX и при расчете дозы и схемы заместительной терапии необходимо учитывать, что введение 1 МЕ FII на 1 кг массы тела пациента повышает активность FII, в среднем, на 2% (восстановление активности FII — тест восстановления = 2). Период полувыведения FII составляет около 60 часов. Таким образом, стандартная терапевтическая доза КПК 20-30 МЕ/кг массы тела пациента (здесь и далее расчет по FIX) повышает активность плазменного FII до 40-60%. У пациентов с активным кровотечением этот показатель может быть меньше.
При тяжелых кровотечениях или больших хирургических вмешательствах у пациентов с дефицитом FII КПК назначается в стартовой насыщающей дозе 20 — 60 МЕ/кг массы тела пациента с последующими повторными инфузиями препарата в дозе 10 — 30 МЕ/кг массы тела пациента с интервалами в 24 — 48 часов для достижения и поддержания активности FII > 20%.
Профилактическое лечение пациентам с дефицитом FII назначается с целью предотвращения развития геморрагического синдрома при наличии персонального или семейного анамнеза тяжелого клинического фенотипа заболевания или при снижении активности FII < 1%. КПК вводится в дозе 20 - 40 МЕ/кг массы тела пациента с интервалом в 5 - 7 дней с целью достижения активности FII ≥ 10% [13].
Сведений о возможном возникновении аллоантител к FII не имеется.
В случае недоступности КПК возможно использование карантинизированной СЗП в дозе 15-25 мл/кг массы тела пациента, повышающей плазменную активность FII до 30-40%. Использование СЗП возможно в исключительных случаях и не должно являться постоянной практикой.
Для купирования легких кровотечений или в случае проведения малого хирургического вмешательства у пациентов с дефицитом FII назначение транексамовой кислоты в дозе 15-20 мг/кг массы тела пациента или 1,0 г х 4 раза в сутки.
При гипопроконвертинемии для купирования или предупреждения кровотечений рекомендуется проведение специфической заместительной терапии следующими препаратами: плазматическим концентратом FVII (неактивированным), эптаког альфа (активированным) – рекомбинантным активированным FVII (rFVIIа), антиингибиторный коагулянтный комплекс (АИКК) и КПК, содержащими FVII [14].
Гемостатическая терапия концентратом плазматического FVII проводится при развитии кровотечения/кровоизлияния в дозе 20-30 МЕ/кг массы тела пациента каждые 4-6 часов до купирования геморрагического синдрома. С целью предупреждения возникновения кровотечения возможно проведение заместительной терапии концентратом FVII в дозе 10-30 МЕ/кг массы тела пациента 3 раза в неделю.

Таблица 3. Терапия РНСК по требованию
Альтернативой концентрату плазматического FVII для лечения пациентов с гипопроконвертинемией является rFVIIа. Для остановки легкого кровотечения препарат вводится однократно в дозе 30-50 мкг/кг массы тела пациента. При возникновении умеренного или тяжелого кровотечения, а также в случае высокого риска развития геморрагических осложнений во время и после проведения хирургических вмешательств рекомендуются повторные введения эптаког альфа (активированного) в дозе 15-30 мкг/кг массы тела пациента с интервалом в 4-6 часов (обычно не менее 3-х инфузий) [2].
При выполнении малых хирургических вмешательств или инвазивных процедур минимальная доза эптаког альфа (активированного) должна составлять 15 мкг/кг массы тела пациента до проведения манипуляции и вводится, как минимум, дважды после окончания процедуры с интервалом в 4-6 часов.
При проведении хирургического лечения частота введения препаратов определяется объемом хирургического вмешательства. Рекомендуется контролировать ПВ не реже 1 раза в 12 часов и проводить заместительную терапию при снижении МНО менее 1,3.
Назначение профилактического режима введения препаратов FVII больным с гипопроконвертинемией рекомендуется при повторных кровоизлияниях в суставы, рецидивирующих кровоизлияниях и кровотечениях другой локализации, при условии, что геморрагический синдром определяет прогноз для здоровья и жизни пациента и значимо нарушает качество его жизни [15].
Частота введений препаратов подбирается индивидуально с учетом клинической картины заболевания. Профилактическое введение препаратов может быть как краткосрочным, (например, при маточных кровотечениях: rFVIIa в дозе 20-40 мкг/кг массы тела пациентки 3-4 инфузии с интервалом в 12-24 часов до достижения клинического ответа), так и длительным (например, при рецидивирующих гемартрозах — введение плазматического концентрата FVII в дозе 20-30 МЕ/кг массы тела пациента 3 раза в неделю).
Таблица 4. Профилактическая терапия РНСК

Новорожденным с гипопроконвертинемией, не имеющих семейного анамнеза тяжелых кровотечений, но с активностью FVII ≤ 1% рекомендуется проведение краткосрочной профилактики эптаког альфа (активированным) в дозе 20-40 мкг/кг массы тела пациента 3 раза в неделю до достижения 6-12- месячного возраста.
Эффективность терапии концентратом FVII и эптаког альфа (активированным) идентична. Имеются сведения о возможном появлении аллоантител к FVII при проведении заместительной терапии. При условии равной эффективности и удовлетворительной индивидуальной переносимости выбор лекарственного средства для заместительной терапии зависит от возможности бесперебойного обеспечения пациента этим препаратом.
В случае отсутствия rFVIIa и концентрата плазматического FVII для остановки кровотечения при дефиците FVII рекомендуется однократно использовать АИКК в дозе 20-40 Ед/кг массы тела однократно в сутки до полной остановки кровотечения или КПК, содержащий FVII, в дозе 20-30 МЕ/кг (FIX) массы тела пациента [8]. В связи с повышенным риском развития тромбозов при использовании этих препаратов повторная инъекция возможна не ранее 24 часов после предыдущего введения препарата.
В случае недоступности rFVIIa, концентрата плазматического FVII, КПК или АИКК возможно назначение карантинизированной СЗП в дозе 15-25 мл/кг массы тела пациента. Однако, использование СЗП возможно в исключительных случаях и не должно являться постоянной практикой.
При гипопроконвертинемии для купирования легких кровотечений или в случае проведения малого хирургического вмешательства с высоким риском развития кровотечения, а также во всех случаях низкого риска развития послеоперационного геморрагического синдрома рекомендуется монотерапия транексамовой кислотой в дозе 15-20 мг/кг массы тела пациента или 1,0 г х 4 раза в сутки [16].
Для лечения пациентов с дефицитом FX рекомендована специфическая заместительная терапия КПК.
Рекомендуется использование плазматических очищенных вирусинактивированных КПК. При назначении КПК необходимо отдельно уточнять содержание FХ, которое указывается в инструкции. КПК содержат FIX и FX приблизительно в эквивалентной активности. При введении КПК в дозе 1 МЕ/кг массы тела пациента активность FX повышается, в среднем, на 2% (восстановление активности FX – тест восстановления = 2). Таким образом, стандартная терапевтическая доза КПК 20-30 (FIX) МЕ/кг массы тела пациента повышает активность плазменного FX до 40-60% [17].
При тяжелых кровотечениях или больших хирургических вмешательствах у пациентов с дефицитом FX КПК назначаются в стартовой насыщающей дозе 20-30 (FIX) МЕ/кг массы тела пациента с последующими повторными инфузиями препарата в дозе 10-20 (FIX) МЕ/кг массы тела пациента с интервалами в 24-48 часов для достижения и поддержания активности FX > 20%.
При дефиците FX долговременное профилактическое лечение рекомендуется с целью предотвращения развития геморрагического синдрома при наличии персонального или семейного анамнеза тяжелого клинического фенотипа предшествующих кровотечений или при снижении активности FX < 2% [11, 16, 17, 27, 28, 36, 40, 41]. КПК назначается в дозе 20-30 (FIX) МЕ/кг массы тела пациента 2-3 раза в неделю с целью поддержания активности FX >1% у взрослых и > 2% у детей. Альтернативный вариант проведения профилактического лечения может быть использован у пациентов с базовой активностью FX < 5%: КПК* в дозе 20-70 (FIX) МЕ/кг массы тела пациента вводится 1 раз в неделю [18, 19].
Сведений о возможном возникновении изоантител к FX не имеется.
В случае недоступности КПК возможно использование карантинизированной СЗП в дозе 15-25 мл/кг массы тела пациента, повышающей плазменную активность FX до 30-40% [27, 41], но это возможно в исключительных случаях и не должно являться постоянной практикой.
Для купирования легких кровотечений или в случае проведения малого хирургического вмешательства пациентам с дефицитом FX рекомендовано проведение монотерапии транексамовой кислотой в дозе 15-20 мг/кг массы тела пациента или 1,0 г х 4 раза в сутки [16, 18].
У женщин с РНСК с рецидивирующими тяжелыми маточными кровотечениями возможно назначение ЗГТ.
Стоматология
Для больных с РНСК важно соблюдение гигиены полости рта, что помогает предотвратить развитие пародонтоза и кариеса. Для чистки зубов необходимо использовать мягкую зубную щетку. Плановые стоматологические осмотры должны проводиться ежегодно. Обычные осмотры стоматолога и чистка зубов могут проводиться без заместительной терапии факторами. При оказании стоматологической помощи важно тесное взаимодействие хирурга-стоматолога и врача гематолога. Удаление зуба или хирургические процедуры должны выполняться под строгим контролем гемостаза и после консультации гематолога.
При стоматологических манипуляциях местная анестезия у пациентов с тяжелыми и среднетяжелыми формами дефицитов FII, FVII, FX должна проводиться только после введения специфического концентрата фактора свертывания [2, 20].
При проведении стоматологических процедур возможно применение транексамовой или других антифибринолитических препаратов с целью уменьшения необходимости в заместительной терапии концентратом фактора. Возможно использование местных гемостатических препаратов после удаления зубов.
При обширных стоматологических процедурах (наложение швов, множественная экстракция зубов) может понадобиться госпитализация пациента в стационар.
Диспансерное наблюдение
Всех пациентов с РНСК рекомендовано регистрировать и наблюдать в специализированном центре. У пациентов или врачей, к которым они обращаются, должна быть постоянная возможность контакта с гематологом, имеющим опыт лечения больных с нарушениями гемостаза. Ведение и лечение пациентов с РНСК проводится группой специалистов различного профиля, включающей гематолога, педиатра, ортопеда, стоматолога, физиотерапевта, врача ЛФК, психолога, имеющих опыт работы с больными гемофилией.
Осмотр пациентов гематологом, ортопедом и стоматологом проводить не менее 2-х раз в год; остальными специалистами — по необходимости [2, 12].
Целесообразно проведение диспансеризации пациентов 1 раз в год в специализированном центре нарушений гемостаза, если центр располагает достаточной клинико-лабораторной базой.
Диспансерное наблюдение за пациентами с РНСК должно включать [20]:
- обязательный динамический мониторинг состояния пациента с оценкой наличия нежелательных явлений при проведении заместительной терапии: появление ингибитора к факторам свертывания крови, индивидуальная непереносимость препаратов, вирусная контаминация;
- оценку изменения психологического или социального статуса пациента;
- оценку состояния периферической венозной системы;
- лечение осложнений РНСК: коррекция дефицита железа, ингибиторов;
- выявление и лечение сопутствующих заболеваний, особенно заболеваний зубов, полости рта, ЖКТ, ЛОР-органов, патологии сердечно-сосудистой системы и др.
Особенности РНСК у детей
Проявлением дефицита FII у детей могут быть пупочные кровотечения или кровоизлияния в ЦНС. Важно помнить, что у детей в первом полугодии жизни нормальная активность FII составляет 26—70%. Значений у взрослых активность FII достигает к шестимесячному возрасту. После достижения возраста 6 мес необходимо проведение неоднократных повторных обследований. Поэтому точно поставить диагноз дефицита FII можно у детей старше 1 года. В любом случае при обнаружении удлинения ПВ у детей в этом возрасте показано назначение препаратов витамина K [4].
У новорожденных с дефицитом FVII существует опасность спонтанных кровоизлияний в ЦНС или пупочных кровотечений. Другие проявления геморрагического синдрома нехарактерны. Физиологическая активность FVII при рождении составляет 28—104% и достигает нормальных значений к шестимесячному возрасту. Поэтому окончательный диагноз устанавливается после достижения ребенком 6—12 месяцев.
У новорожденных с дефицитом FX возможны кровоизлияния в ЦНС или пупочные кровотечения. Физиологическая активность FX при рождении составляет 12—68% и повышается к шестимесячному возрасту. Поэтому окончательное установление диагноза дефицита FX у новорожденных требует обязательного сравнения результатов лабораторного исследования с референсными интервалами допустимых в неонатальном периоде значений и повторного обследования после достижения шестимесячного возраста.
При выявлении у детей первого полугодия жизни сниженной активности FХ показано назначение препаратов витамина K [21].
Ведение женщин с РНСК во время беременности и родом
В течение нормально протекающей беременности активность FII существенно не меняется и обычно остается недостаточной для неосложненного родоразрешения у женщин с тяжелым дефицитом протромбина. При активности FII < 20% для предотвращения геморрагических осложнений в родах необходимо введение КПК в дозе 20—40 МЕ/кг однократно при начале родовой деятельности или перед кесаревым сечением для достижения активности FII, составляющей 20—40%. Повторные инфузии препарата проводятся с интервалом в 48 часов в дозе 10—20 МЕ/кг с целью поддержания активности FII >20% в течение как минимум трех дней после родов. При отсутствии клинических проявлений заболевания во время беременности заместительная терапия КПК не проводится, в случае развития кровотечения терапия проводится по общим принципам [22].
Активность FVII в течение нормально протекающей беременности повышается. Женщины с легким дефицитом FVII могут достигнуть к моменту родов необходимого гемостатического уровня активности FVII без проведения специфической заместительной терапии. У беременных с тяжелым дефицитом FVII есть риск геморрагических осложнений в родах. Женщинам с активностью FVII 20% перед кесаревым сечением или с началом родовой деятельности необходимо проведение заместительной терапии препаратом Эптаког альфа (активированным) в дозе 15—30 мкг/кг каждые 4—6 часов в течение не менее 3-х дней. Остальным женщинам с дефицитом FVII Эптаког альфа назначается только при развитии кровотечения согласно общим принципам терапии [23, 24].
У женщин с активностью FX менее 30% возможны проблемы с зачатием. Беременность осложняется ранними выкидышами в первом триместре, геморрагическими проявлениями (чаще геморрагическим циститом) во втором и третьем триместрах. Необходимый уровень FX до и во время беременности должен быть не менее 30%.Несмотря на физиологическое повышение активности FX в течение нормально протекающей беременности, у женщин с тяжелым дефицитом FX его уровень к родам обычно остается недостаточным для обеспечения нормального гемостаза. Женщинам с активностью FX 30% в третьем триместре беременности с наличием в анамнезе кровотечений до беременности, а также перед кесаревым сечением или с началом родовой деятельности необходима заместительная терапия КПК в дозе 20—40 МЕ/кг для достижения активности FX > 40%. Дальнейшие инфузии КПК проводятся с интервалом в 24 часа в дозе 10—20 МЕ/кг в течение как минимум трех дней для поддержания активности FX не менее 30% [25].
Вакцинация.
Пациенты с РНСК могут быть вакцинированы. Особенно важно проведение вакцинации от гепатита В. При вакцинации предпочтение отдается оральному или подкожному введению препарата, по сравнению с внутримышечным или внутрикожным. Если для данной вакцины доступен только внутримышечный путь введения, необходима заместительная терапия для предотвращения развития гематомы. В этом случае заместительную терапию проводят накануне вакцинации. В день вакцинации введение препарата не рекомендуется. Нельзя проводить вакцинацию во время кровотечения [20].
Нежелательная медикаментозная терапия.
Больным с РНСК не рекомендовано применение препаратов, ухудшающих функцию тромбоцитов или свертывания крови. Применение таких препаратов может привести к развитию тяжелых кровотечений, которые не купируются введением концентратов факторов свертывания крови. Однако развившийся тромбоз может потребовать применения антикоагулянтов. Предпочтение надо отдавать препаратам кратковременного действия. Каждый раз необходимо анализировать соотношение пользы и риска от применения антикоагулянтов и антиагрегантов [12].
Проведение лабораторных исследований.
Лабораторные анализы лежат в основе диагностики и контроля у пациентов с РНСК. Требования к условиям и технике отбора образцов и выполнения исследований не отличаются от стандартных. Важным аспектом лабораторных исследований является участие в системе контроля качества. При диагностике РНСК оптимально участвовать не только в государственной, но и в международной системе контроля качества лабораторных исследований, охватывающей основные коагулологические параметры [3, 8, 12].
Обучение пациентов и членов их семей.
Обучение пациентов и членов их семей – необходимое условие обеспечения адекватной помощи больным с РНСК. Обучение начинается сразу после установления диагноза и проводится на постоянной основе врачами и медицинскими сестрами центра, в котором наблюдается пациент. Обучение проводиться индивидуально при посещении центра и в рамках школы пациента с РНСК.
Основные направления обучения пациента и членов его семьи:
- что такое РНСК,
- особенности заболевания в детском возрасте,
- навыки оценки состояния ребенка,
- навыки оценки симптомов, характера и тяжести кровотечения,
- хранение и использование концентратов факторов свертывания крови,
- показания и дозы заместительной терапии,
- навыки проведения инфузии в домашних условиях,
- уход за венами,
- применение других гемостатических препаратов,
- физическая активность,
- психологическая и социальная адаптация,
- профессиональная ориентация,
- юридические аспекты.
Алгоритм ведения пациентов с РНСК


Медицинская реабилитация
Реабилитация
Пациентам с РНСК после кровоизлияния в ЦНС, с поражением опорно-двигательного аппарата рекомендовано санаторно-курортное лечение. Разработка реабилитационных мероприятий должна проводиться совместно специалистами по реабилитации, курортологии и гематологами, имеющими опыт лечения пациентов с нарушениями свертывания крови. Лечение можно проводить в санаторно-курортных организациях в климатической зоне проживания пациента, а также на бальнеологических курортах.
Целесообразно проведение школ психологической адаптации для пациентов с РНСК.
Информация
Источники и литература
- Клинические рекомендации Национального гематологического общества
- 1. Acharya SS, Coughli A, Dimichele DM et al. Rare Bleeding Disorder Registry: deficiencies of factors II, V, VII, X, XIII, fibrinogen and dysfibrinogenemias. J Thromb Haemost 2004; 2:248—256. 2. Bolton-Maggs PH, Perry DJ, Chalmers EA et al. The rare coagulation disorders — review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ Organisation. Haemophilia 2004; 10:593—628. 3. Момот А. П. Принципы и алгоритмы клинико-лабораторной диагностики. — СПб: ФормаТ, 2006. 4. Girolami A, Scarano L, Saggiorato G et al. Congenital deficiencies and abnormalities of prothrombin. Blood Coagulation & Fibrinolysis 1998; 9:557—569. 5. Di Minno MN, Dolce A, Mariani G et al. Bleeding symptoms at disease presentation and prediction of ensuing bleeding in inherited FVII deficiency. Thromb Haemost 2013; 109:1051—1059. 6. Bernardi F, Dolce A, Pinotti M et al. Major differences in bleeding symptoms between factor VII deficiency and hemophilia B. J Thromb Haemost 2009; 7:774—779. 7. Brown DL, Kouides PA. Diagnosis and treatment of inherited factor X deficiency. Haemophilia 2008; 14:1176—1182. 8. Баркаган З. С., Момот А. П. Диагностика и контролируемая терапия нарушений гемостаза. — М. Ньюдиамид, 2001. 9. Шабалов Н. П. Гемостаз в динамике первой недели жизни как отражение механизмов адаптации к внеутробной жизни новорожденных. Педиатрия 2000; 3:84—91. 10. Руководство по гематологии в 3 томах / Под ред. А. И. Воробьева. — М.: Ньюдиамед, 2005. 11. Основы клинической гемостазиологии и гемореологии: монография / Под ред. И. Л. Давыдкина, А. П. Момота, Н. И. Зозули, Е. В. Ройтмана. — Самара: ООО ИПК «Самарская Губерния», 2017. 12. Mamford AD, Ackroyd S, Alikhan R et al. Guideline for the diagnosis and management of the rare coagulation disorders. Bri J Haematol 2014; 5:1—23. 13. Lancellotti S, Basso M, De Cristofaro R. Congenital prothrombin deficiency: an update. Seminars in Thrombosis and Hemostasis 2013; 39:596—606. 14. Di Paola J, Nugent D, Young G. Current therapy for rare factor deficiencies. Haemophilia 2001; 7:16—22. 15. Napolitano M, Giansily-Blaizot M, Dolce A et al. Prophylaxis in congenital factor VII deficiency: indications, efficacy and safety. Results from the Seven Treatment Evaluation Registry (STER). Haematologica 2013; 98:538—544. 16. Mannucci PM, Duga S, Peyvandi F. Recessively inherited coagulation disorders. Blood 2004; 104:1243—1252. 17. Kouides PA, Kulzer L. Prophylactic treatment of severe factor X deficiency with prothrombin complex concentrate. Haemophilia 2001; 7:220—223. 18. McMahon C, Smith J, Goonan C et al. The role of primary prophylactic factor replacement therapy in children with severe factor X deficiency. Br J Haematol 2002; 119:789—791. 19. Bowles L, Baker K, Khair K et al. Prophylaxis with prothrombin complex concentrate in four children with severe congenital factor X deficiency. Haemophilia 2009; 15:401—403 20. Keeling D, Tait C, Makris M. Guideline on the selection and use of therapeutic products to treat haemophilia and other hereditary bleeding disorders. A United Kingdom Haemophilia Center Doctors’ Organisation (UKHCDO) guideline approved by the British Committee for Standards in Haematology. Haemophilia 2008; 14:671—684. 21. Peyvandi F, Palla R, Menegatti M et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost 2012; 10:615—621. 22. Kadir R, Chi C, Bolton Maggs P. Pregnancy and rare bleeding disorders. Haemophilia 2009; 15:990—1005.Baumann Kreuziger LM, Morton CT, Reding MT. 23. Is prophylaxis required for delivery in women with factor VII deficiency? Haemophilia 2013; 19:827—832. 24. Kulkarni AA, Lee CA. Kadir RA. Pregnancy in women with congenital factor VII deficiency. Haemophilia 2006; 12:413—416.Beksac MS, Atak Z, Ozlu T. 25. Severe factor X deficiency in a twin pregnancy. Archives of Gynecology and Obstetrics 2010; 281:151—152.
Редкие болезни: как распознать наличие орфанного заболевания и как лечить

Заболевания бывают острые и хронические. Некоторыми заболеваниями переболевает практически каждый человек в течение своей жизни. Другие же болезни встречаются очень редко. Какие бывают редкие заболевания у людей? Как помочь пациентам с редкими заболеваниями?
Редкие болезни
К редким болезням относятся те заболевания, которые встречаются у небольшого количества населения. Их еще называют орфанными. При этом термин «редкие орфанные заболевания» не является правильным, потому что непривычное слово «орфанные» как раз и означает «редкие». Единого термина для орфанных патологических состояний до сих пор нет.
Существует ли единый список редких болезней? Нет, такой перечень редких заболеваний не разработан, потому что распространенность различных заболеваний отличается в разных популяциях. То есть, в одних странах какая-то конкретная патология практически не встречается и входит в список самых редких, а в другой регистрируется довольно-таки часто.
По данным медицинского сообщества Соединенных Штатов Америки к редким патологиям относят те болезни, которые зарегистрированы менее чем у 200 тысяч людей, то есть, примерно у одного человека из 1500. В Японии редких патологий выделено меньше, потому что к редким относятся те, которые отмечаются у 1 из 2500 человек. Ими страдают меньше 50 тысяч японцев. По европейским статистическим данным в совокупности редкими болезнями страдают менее 10 процентов жителей.
На территории нашей страны редкими числятся те патологии, которые встречаются не более чем у 10 человек из 100 тысяч. Минздравсоцразвития России изначально выделял 86 редких патологий. Сейчас этот список расширен до 215 болезней. Столь значимое расширение списка орфанных болезней произошло в 2014 году 7 мая. Хотя по миру насчитывается редких болезней гораздо больше (более пяти тысяч).
Таким образом, по разным литературным источникам к редким относятся патологии, уровень распространенности которых от 1 на 1000 до 1 на 200000 населения.
В феврале в мире отмечают Международный день редких болезней. Дата выбрана символично: 29 февраля, ведь такое число бывает не каждый год.

Каковы ваши шансы достичь долголетия: пройдите тест от ученых
Редкие генетические синдромы и врожденные аномалии

Многие генетические патологии передаются из поколения в поколения, однако среди них много редких. В нашей стране в перечне редких болезней их насчитывается более 20. К ним относятся ахондроплазия, несовершенный остеогенез, редкие заболевания кожи (врожденный ихтиоз, буллезный эпидермолиз, различные факоматозы), туберозный склероз, болезни Марфана и кошачьего крика, VATER-синдром, синдромы Нунан, Гольденхара, Блума, Беквита-Видемана и др.
Больные генетическими заболеваниями требуют наблюдения у генетика и врачей других специальностей. Вылечиться от данных болезней не представляется возможным.
Редкие заболевания крови и иммунной системы
В списке редких болезней крови нашей страны числится около 50 болезней. К ним относятся редкие виды анемий (наследственный сфероцитоз, серповидно-клеточная и наследственная сидеробластная анемии), талассемии, гемоглобинопатии, разные типы красноклеточной аплазии, нарушения свертываемости крови (гемофилии, болезнь Виллебранда, врожденные дефициты факторов свертывания, идиопатическая тромбоцитопеническая пурпура), метгемоглобинемии, семейный эритроцитоз, гистиоцитоз Х, различные врожденные иммунодефициты (синдромы Вискотта-Олдрича, Ди Джорджа, Блума, синдром адгезии лейкоцитов) и другие патологии.
Не всегда диагноз ставится в раннем детстве, а в некоторых случаях человек узнает о своей необычности, будучи взрослым. Но в случае тяжелых форм люди с заболеваниями крови и иммунной системы обследуются очень рано. К сожалению, многие пациенты погибают в раннем детстве.
Редкие заболевания легких, сердца и других внутренних органов
Редкие заболевания легких, сердца, мочеполовой системы также представлены в перечне редких болезней нашей страны. Их насчитывается более 20. К таковым относятся следующие аномалии: некоторые формы легочной гипертензии, разные виды кардиомиопатий, врожденные патологии проводящей системы сердца, наследственные формы внезапного пневмоторакса, болезнь Крона, неспецифический язвенный колит, грануломатоз Вегенера, некоторые формы нефротического синдрома, гломерулопатии и другие болезни.
Некоторые болезни внутренних органов можно скорректировать оперативным путем. При других болезнях требуется прием лекарств пожизненно. В случае нетяжелых заболеваний требуется только наблюдение.
Редкие инфекционные болезни
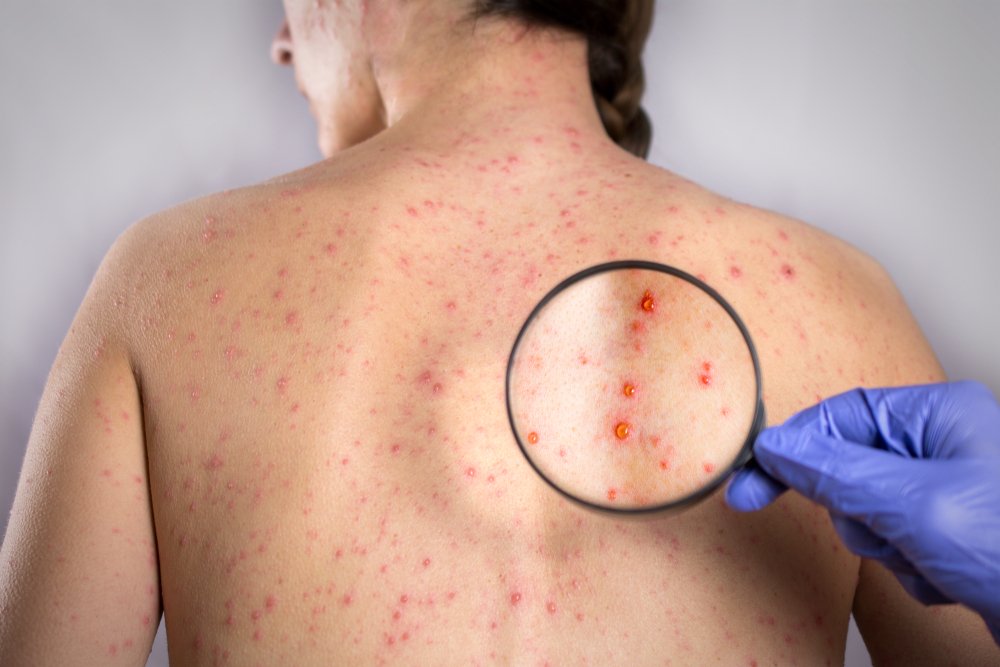
В списке редких патологических состояний и синдромов в нашей стране не числятся инфекционные болезни. Дело в том, что большинством инфекционных болезней можно управлять, что будет влиять на их частоту встречаемости. Например, мы крайне редко встречаемся с полиомиелитом из-за массовой вакцинации, хотя в некоторых странах дети продолжают оставаться инвалидами из-за полиовирусов. Если российские жители начнут массово отказываться от прививок, то полиомиелит и другие инфекционные заболевания очень быстро вернутся и на территорию нашей страны.
Некоторые инфекционные болезни встречаются в одних странах чаще, в других реже из-за особенностей климата и ландшафта, социально-бытовых условий населения и даже уровня медицины. Самые редкие заболевания инфекционного генеза одной страны могут быть обыденными патологиями в других странах (например, малярия и различные тропические лихорадки).
Редкие психические заболевания и болезни нервной системы
В нашей стране выделяется немного редких психических болезней. К ним относятся синдромы Ретта и де ла Туретта. Редких болезней нервной системы насчитывается более 20. К ним относятся различные спинальные мышечные атрофии, миопатии, синдром Луи-Барр, атаксия Фридрейха, прогрессирующий бульбарный паралич у детей, различные дегенеративные болезни, некоторые типы эпилепсии, синдром Гийена-Барре, сирингомиелия и др.
Редкие заболевания нервной системы очень сильно ухудшают качество жизни пациентов. Многие болезни только прогрессируют с годами. К сожалению, остановить этот процесс практически невозможно.
Редкие онкологические болезни
В список редких онкологических болезней нашей страны входят около 40 патологий. К ним относятся такие болезни как саркомы костей и суставных хрящей конечностей, ретинобластома (опухоль сетчатки глаза), болезнь Ходжкина, различные виды лимфолейкозов и миелоидных лейкозов, множественная миелома, различные типы неходжскинской лимфомы, саркоидоз и др. Есть среди редких онкологических болезней и доброкачественные заболевания, например, семейный аденоматозный полипоз кишечника.
Эти заболевания редко являются врожденными, хотя и такое встречается. Чаще человек заболевает ими в течение своей жизни (кто-то в детстве, а кто-то, будучи взрослым). Смертность от злокачественных редких болезней очень высока. Их лечение дорогостоящее и длительное.
Редкие эндокринные болезни и расстройства метаболизма
В перечень редких болезней эндокринной системы и патологий обмена веществ входят такие болезни как врожденный гиперинсулинизм, детская пролактинома, гипопитуитаризм, адреногенитальный синдром, болезнь Иценко-Кушинга, синдром Ларона, гермафродитизм, прогерия, фенилкетонурия, лейциноз, цистиноз, гомоцистинурия, гликогенозы, галактоземия, мукополисахаридозы, гипофасфатемический рахит, муковисцидоз и многие другие нарушения. Всего выделяют около 50 заболеваний, каждое из которых имеет еще разные типы и варианты течения.
Очень важно найти грамотного врача, который сможет разобраться во всех нарушениях обмена гормонов и других веществ и поможет больному. При подозрении на выше перечисленные болезни не обойтись без генетического обследования. Данное обследование важно для выяснения всех тонкостей нарушения обмена.
Неонатальный скрининг: помощь в ранней диагностике

Не все редкие болезни успешно лечатся. Большинство из них являются хроническими, а порой и прогрессирующими. Однако есть такие заболевания, которые при ранней диагностике и своевременном правильном лечении удается держать под контролем. На некоторые из этих болезней обследуют всех детей с самого рождения. Проводят исследование на 4-7 дни жизни (что зависит от того, доношенный ли родился малыш). Такое массовое обследование называется неонатальным скринингом. В каждой стране свой перечень заболеваний для неонатального скрининга.
Дети с редкими болезнями (которые удалось подтвердить с помощью скрининга) начинают получать лечение с первых месяцев жизни. Благодаря этому качество их жизни не сильно отличается от таковой у здоровых сверстников. В нашей стране с рождения всех детей обследуют на 5 болезней и выясняют очень рано, нет ли у них муковисцидоза, фенилкетонурии, галактоземии, адреногенитального синдрома, врожденного гипотиреоза. Да, эти редко встречающиеся болезни нельзя полностью вылечить, всю жизнь приходится принимать определенные лекарства.
Лечение редких болезней
Учитывая тот факт, что многие редко встречающиеся патологии являются генетическими, полностью вылечить их невозможно. Некоторые редкие болезни проявляются с раннего детства тяжелыми нарушениями, поэтому почти треть больных детей не доживает до пятилетнего возраста.
Наблюдением и лечением больных с редкими патологиями занимаются врачи разных специальностей. Все зависит от того, какие органы и системы страдают у больного. При некоторых генетических синдромах у больных имеется целый комплекс проблем со стороны многих органов и систем, поэтому их наблюдают врачи разных специальностей.
Разработан Закон об орфанных препаратах в нашей стране. Его разработали для того, чтобы ученые продолжали исследования редких патологий и разрабатывали лекарственные средства для улучшения качества жизни больных.
На сегодняшний день в Российской Федерации есть утвержденный перечень орфанных патологий, состоящий из 24 наименований заболеваний. В борьбе с этими болезнями разработано лечение. Если его не проводить, то высок риск снижения продолжительности жизни и формирования инвалидности. Все пациенты, имеющие диагноз из данного списка занесены в регистр пациентов с орфанными патологиями.
Проблема лечения редких патологий в том, что большинство препаратов очень дорого стоят, требуются они в большом количестве, а многие препараты и вовсе не прошли регистрацию в нашей стране и пока не могут быть закуплены для больных. Как правило, лекарства людям с редкими патологиями нужно принимать почти постоянно, некоторым требуются хирургические операции и трансплантации донорских органов.
Сейчас в Российской Федерации реализуется программа «7 нозологий». Больные, имеющие одно из этих семи болезней, получают бесплатное лечение. Медикаменты для них покупаются за счет бюджетных средств. Для кого же они выделяются? Для людей, страдающих гемофилией, перенесшим трансплантацию органов и тканей, страдающим болезнью Гоше, больным рассеянным склерозом, а также муковисцидозом, гипофизарным нанизмом, злокачественными патологиями крови и лимфоидной ткани.
На современном этапе в России более 60% людей с орфанными патологиями получают лечение. Некоторые пациенты в терапии не нуждаются, им требуется просто наблюдение.
Профилактика редких болезней
Самые редкие заболевания являются большой проблемой, как в медико-социальном, так и в экономическом плане. Реально ли предупредить редкое заболевание? К сожалению, профилактических мер в отношении возникновения редких болезней не так много, как хотелось бы. Но все же можно выделить некоторые звенья профилактики:
- Грамотное планирование беременности;
- Изучение своей родословной, чтобы избежать родственных браков (среди них чаще выявляются наследственные заболевания);
- Проведение скринингов при беременности, которые могут указать на риск формирования редкой патологии; прерывание беременностей по желанию родителей, если у плода выявлено тяжелое редкое генетическое заболевание или порок развития.
Использованы фотоматериалы Shutterstock
Читайте далее

Где можно сдать анализ на гепатит
Можно ли обследоваться на наличие гепатита бесплатно? Какие анализы и где можно сдавать, какие нужны направления и подготовка.

Какие болезни диагностируют по слюне?
Как проводят диагностику по слюне, какие болезни можно так обнаружить, и почему это лучше анализа крови — в статье MedAboutMe

Какие проблемы с кожей помогут решить средства с гиалуроновой кислотой?
Сухость кожи, чувство стянутости — всё это и другие проблемы, которые решают средства с гиалуроновой кислотой.
Кровь редкие болезни крови
Takeda стремится преобразовать лечение редких заболеваний в иммунологии, гематологии, нарушений обмена веществ и лизосомальных накоплений. Симптомы редких генетических и метаболических заболеваний сильно различаются и прогрессируют по-разному от человека к человеку, что означает, что людям, страдающим этим заболеванием, часто неправильно ставят диагноз.
Редкие заболевания крови
Takeda — лидер в области редких гематологических заболеваний самым длительным опытом и лидирующим на рынке портфелем препаратов, подкрепленным установленными профилями и эффективностью с многолетним практическим опытом.
Более 70 лет мы внедряем инновации для пациентов, обладаем широким спектром вариантов лечения по всем показмания в гемофилии. Наш опыт лидеров в области гематологии означает, что мы хорошо подготовлены к удовлетворению сегодняшних разработанных потребностей, следуя будущим лечениям свертываемости крови. Вместе с гематологическим сообществом мы надеемся на будущее, включая более раннюю диагностику, более раннюю и персональную защиту от кровотечений и более раннюю защиту от кровотечения за пациентом.
Заболевания
Гемофилия
Гемофилия — это нарушение свертываемости крови, которое в основном поражает мужчин. Люди с гемофилией испытывают недостаток, называемых факторами свертывания, поэтому их сверты должным образом. По оценкам, более 400 000 человек во всем мире живут с гемофилией.
Болезнь Виллебранда
Болезнь Виллебранда (БВ), одно из наиболее распространенных наследственных нарушений свертываемости крови, вызвано дефицитом или дисфункцией фактора фон Виллебранда, когда не может образоваться нормальный тромб или на это требуется больше времени, что приводит к длительному кровотечению. БВ встречается примерно у 1% населения мира, включая 3,2 миллиона человек только в США. Девять из 10 человек с ВБ не диагностированы.
Подробный доклад

Новые решения для достижения Персонализированного ухода за Пациентами, Живущими с Редкими заболеваниями
Компания Takeda в сотрудничестве с компанией PwC опубликовала доклад «Ми ссия: ориентированность на пациента — к ак технологии могут помочь достичь персонализированного ухода за пациентом с редкими заболеваниями» , чтобы обеспечить надежность постоянного развития возможностей в одном из главных приоритетов в области исследований и исследований. Такеда. ПОДРОБНЕЙ
Редкие болезни иммунной системы
В области редких заболеваний иммунологии мы фокусируемся на продвижении непрерывных инноваций и персонализированной помощи через наш портфель плазменных продуктов и инновационных целевых методов лечения, устройств, диагностики и других технологических услуг для пациентов с редкими иммунологическими нарушениями. Наши основные терапевтические направления включают Наследственный ангионевротический отек, иммунодефицитные состояния, редкие аутоиммунные расстройства, посттрансплантационные осложнения и редкую специализированную помощь.
Заболевания
Наследственный ангионевротический отек (HAО)
НАО — редкое генетическое заболевание, характеризующееся высокими приступами отека в различных частях тела, включая живот, лицо, грудь, гениталии, руки и гортань. Отек может быть изнурительным и болезненным 1,2,3 . Приступы, которые закрывают дыхательные пути, могут вызвать удушье и первое опасны для жизни 2,3 . HAО поражает примерно от 1 из 10 000 до 1 из 50 000 человек во время. Это заболевание часто не распознают, не диагностируют и не лечат 1,4 .
Иммунодефицитные Заболевания
Первичные иммунодефициты (ПИД)
Первичные иммунодефициты (ПИД) — это группа из более чем 350 редких заболеваний, которые вызывают иммунную систему, не функционирующую должным образом 5,6 . Люди с ПИД могут быть более восприимчивы к болезням, восстанавливаются и имеют повторяющиеся инфекции 5 , такие как синусит, бронхит, пневмония или желудочно-кишечные инфекции 6 . Врачи иногда лечат инфекции, не замеченные основные причины, уязвимыми поврежденными жизненно важными органами, физической инвалидности или даже повышенному риску смертности 7,8 .
Вторичный иммунодефицит (ВИД)
Вторичные иммунодефициты являются следствием таких состояний, как хронический лимфолейкоз и множественная миелома. Они могут привести к повреждениям органов-мишеней и смерти.
Редкие аутоиммунные расстройства
Нервно-мышечные заболевания, при которых происходит нарушение регуляции иммунной системы, приводит к атаке ткани хозяина, часто со сложными механизмами заболеваний. Мультифокальная моторная нейропатия (ММН) — это хроническая нейропатия, которая вызывает прогрессирующую слабость конечностей без объективной сенсорной потери. Синдром Гийена Барре (СГБ) — это угрожающая жизни нейропатия, характеризующаяся прогрессирующей слабостью конечностей и онемением дыхательных, лицевых и глотательных мышц.
Хроническая воспалительная демиелинизирующая полинейропатия (ХВДП) — это заболевание периферических функций, характеризующееся прогрессирующей нервной слабостью конечностей и нарушением сенсорной функции конечностей.
Гипоальбуминемия и Гиповолемия
Гипоальбуминемия возникает при аномально низком уровне альбумина в крови. Это распространенная проблема среди людей с острыми и хроническими заболеваниями, включая ожоги и респираторный дистресс-синдром взрослых. Гиполеволемия из-за уменьшения объема крови или кислорода. Симптомы включают слабость, усталость, обморок и головокружение.
Тяжелый врожденный дефицит протеина С (ТВДПС)
Тяжелый врожденный дефицит протеина — это редкое аутосомно-рецессивное заболевание, которое в основном встречается у новорожденных. Одним из основных симптомов является молниеносная пурпура — острое, часто смертельное тромботическое заболевание, проявляющееся в виде пятен крови, кровоподтеков и изменение цвета кожи в результате коагуляции в мелких кровеносных сосудах внутри кожи и быстро приводит к некрозу кожи и диссеминированному внутрисосудистому свертыванию. Возраст появления первых симптомов может составлять от нескольких часов до 2 недель.
Дефицит протромбина
Дефицит протромбина — это нарушение свертываемости крови, связанное с недостатком витамина К, замедляет процесс свертывания крови. Люди с этим заболеванием часто испытывают длительное кровотечение после травмы или операции.
Дефицит альфа-1 антитрипсина (ААТ или дефицит Альфа-1)
Дефицит альфа-1 антитрипсина (ААТ или дефицит Альфа-1) -это заболевание, которое приводит к снижению уровня белка альфа-1 антитрипсина (ААТ) в крови и легких. Этот белок, который вырабатывает, в основном, безопасные ткани от химических веществ, выделяемых лейкоцитами 9 .
Редкие заболевания обмена веществ
У Takeda сильное наследие в разработке методов лечения лизосом накоплений, с портфелем, который включает коммерческие продукты, поздние стадии исследовательской терапии и кандидатов на производство. Доступны медицинские и исследовательские организации, способствующие глобальному развитию и генетике. Мы стремимся помочь сократить время между появлением симптомов и постановкой диагноза и ускорить редактирование новых инновационных методов лечения.
Заболевания
Синдром Хантера
Синдром Хантера, или мукополисахаридоз II (МПС II), является серьезным генетическим заболеванием, которое в первую очередь поражает мужчин. Он препятствует способности организма расщеплять и перерабатывать специфические мукополисахариды, также известные как гликозаминогликаны. Синдром Хантера является одним из нескольких родственных заболеваний лизосомального накопления.
Болезнь Фабри
Болезнь Фабри — это расстройство лизосомального накопления, которое нарушает способность организма расщеплять определенное жирное вещество (глоботриаозилцерамид или Gb3). Предположительно, данным заболеванием поражено от 8 000 до 10 000 человек во всем мире, как мужчин, так и женщин.
Болезнь Гоше
Болезнь Гоше — это редкое наследственное заболевание, наиболее распространенное в семействе редких заболеваний, как нарушения лизосомального накопления. Предположительно болезнь Гоше поражает 1 на 100 000 человек в мире и 1 на 855 человек в еврейской общине ашкенази 10 . Пациенты типа с болезнью Гоше 1 имеют симптомы и заболевания, что затрудняет диагностику.
Гипопаратиреоз
Гипопаратиреоз — это редкое эндокринное заболевание, вызывающее повреждение паращитовидных желез в результате хирургического вмешательства, аутоиммунного заболевания или генетического нарушения. В результате этого средства не способны вырабатывать кровь паратиреоидного гормона (ПТГ), который важен для контроля уровня кальция и витамина D в. Недостаточное количество ПТГ может нарушить важные функции некоторых органов в организме, включая почки.
Ссылки :
- Cicardi M, Bork K, Caballero T. и др. От имени HAWK (Международная рабочая группа по наследственному ангионевротическому отеку). Доказательные рекомендации по терапевтическому лечению ангионевротического лечения отека недостаточности ингибитора C1: консенсусный отчет Международной рабочей группы. Аллергия. 2012; 67 (2): 147-157.
- Zuraw BL. Наследственный ангионевротический отек. N Engl J Med. 2008; 359 (10): 1027-1036.
- Банерджи А. Бремя болезни у пациентов с наследственным ангионевротическим отеком. Ann Allergy Asthma Immunol. 2013; 111 (5): 329-336.
- Лонгхерст Х. Дж., Борк К. Наследственный ангионевротический отек: причины, проявления и лечение. Br J Hosp Med. 2006; 67 (12): 654-657.
- Фонд иммунодефицита, «О первичных иммунодефицитах: http://primaryimmune.org/about-primary-immunodeficiencies/ . По состоянию на 18 апреля 2016 г.
- Фонд иммунодефицита, «О первичных иммунодефицитах: инфекции». http://primaryimmune.org/about-primary-immunodeficiencies/relevant-info/infections/ . По состоянию на 18 апреля 2016 г.
- Riedl M, Rumbak M. Clin Pulm Med. 2010; 17 (2): 88-95.
- Резник ES и др. Кровь. 2012; 119 (7): 1650-1657.
- Фонд Альфа-1 «Что такое Альфа-1». Брошюра. Доступно по адресу: https://www.alpha1.org/Alpha1/wp-content/uploads/2019/09/Alpha1Brochure.pdf . По состоянию на декабрь 2019 г.
- GuggenbuhlP, GrosboisB, ChalèsG. Болезнь Гоше. Костный сустав позвоночника. 2008; 75 (2): 116-124.
Источник https://diseases.medelement.com/disease/%D1%80%D0%B5%D0%B4%D0%BA%D0%B8%D0%B5-%D0%BD%D0%B0%D1%80%D1%83%D1%88%D0%B5%D0%BD%D0%B8%D1%8F-%D1%81%D0%B2%D0%B5%D1%80%D1%82%D1%8B%D0%B2%D0%B0%D0%BD%D0%B8%D1%8F-%D0%BA%D1%80%D0%BE%D0%B2%D0%B8-%D0%BD%D0%B0%D1%81%D0%BB%D0%B5%D0%B4%D1%81%D1%82%D0%B2%D0%B5%D0%BD%D0%BD%D1%8B%D0%B9-%D0%B4%D0%B5%D1%84%D0%B8%D1%86%D0%B8%D1%82-%D1%84%D0%B0%D0%BA%D1%82%D0%BE%D1%80%D0%BE%D0%B2-%D1%81%D0%B2%D0%B5%D1%80%D1%82%D1%8B%D0%B2%D0%B0%D0%BD%D0%B8%D1%8F-%D0%BA%D1%80%D0%BE%D0%B2%D0%B8-ii-vii-x-%D0%BA%D1%80-%D1%80%D1%84-2018/16769
Источник https://medaboutme.ru/articles/redkie_bolezni_kak_raspoznat_nalichie_orfannogo_zabolevaniya_i_kak_lechit/
Источник https://www.takeda.com/ru-kz/what-we-do/areas-of-focus/Rare_deseases/