Синдром Гурлера ( Болезнь Пфаундлера-Гурлер , Мукополисахаридоз IH )

Синдром Гурлера – это тяжелое наследственное метаболическое заболевание обмена веществ из группы мукополисахаридозов, характеризующееся избыточным накоплением гликозаминогликанов (ГАГ) в различных органах и тканях, что приводит к их выраженной дисфункции. Клиническая картина крайне разнообразна, включает задержку психомоторного развития, грубые деформации костей черепа и скелета, сердечно-легочные нарушения и пр. Диагностика основана на определении экскреции гликозаминогликанов с мочой и активности фермента альфа-идуронидазы в крови, данных молекулярно-генетических тестов. Лечение заключается в заместительной ферментной терапии.
МКБ-10
E76.0 Мукополисахаридоз, тип I

- Причины синдрома Гурлера
- Патогенез
- Симптомы
- Осложнения
- Диагностика
- Лечение синдрома Гурлера
- Консервативная терапия
- Хирургическое лечение
- Реабилитация
Общие сведения
Синдром Гурлера (болезнь Пфаундлера-Гурлер, мукополисахаридоз IH или первого типа) относится к лизосомным болезням накопления с аутосомно-рецессивным типом наследования, проявляется практически с первых месяцев жизни. Заболевание впервые было описано австрийским врачом-педиатром Гертрудой Гурлер. Патология считается одним из самых распространенных мукополисахаридозов, встречается повсеместно. Синдром Гурлера, по разным данным, выявляется у 1:40 000-1:100 000 новорожденных. Значимые гендерные статистические различия отсутствуют.
Синдром Гурлера
Причины синдрома Гурлера
Возникновение заболевания связано гетерогенными мутациями (мелкими делециями, дефектами сайта сплайсинга) гена IDUA, кодирующего синтез энзима альфа-L-идуронидазы. Ген локализован в коротком плече 4 хромосомы в локусе 4p16.3. Наиболее частые мутации гена – Q70X и W402X. Лизосомальный фермент α-L-идуронидаза регулирует метаболизм основных структурных компонентов межклеточного матрикса соединительной ткани – отвечает за деградацию гликозаминогликанов гепарансульфата и дерматансульфата.
Вследствие генетически детерминированного дефекта фермента происходит избыточное накопление ГАГ в лизосомах клеток. Наиболее значимым фактором риска развития синдрома Гурлера является наличие близкого родственника, страдающего этой болезнью. Если у одного из родителей имеется мутантный ген, вероятность рождения больного ребенка составляет 25%.
Патогенез
В результате нарушения внутрилизосомного гидролиза и последующего накопления межклеточных компонентов соединительной ткани высвобождаются воспалительные медиаторы – оксид азота, фактор некроза опухоли-альфа, развивается дисфункция органов. Поскольку соединительная ткань в той или иной степени входит в состав практически каждого органа, поражение носит мультисистемный характер.
Накопление мукополисахаридов в хрящевой ткани вызывает нарушение роста костей, их грубую деформацию. В стенках сосудов, клапанном аппарате сердца, мозговых оболочках развивается фиброз. При патологоанатомическом исследовании обнаруживается увеличение органов, гиперклеточность, дезорганизация. Отмечается уменьшение количества протеогликанов и коллагеновых волокон.
Симптомы
Клинические признаки заболевания начинают проявляться уже на первом месяце жизни. Иногда с рождения отмечается увеличение селезенки и печени, пупочные, паховые грыжи. Ярко выражены симптомы поражения опорно-двигательного аппарата. Достаточно быстро развиваются контрактуры и тугоподвижность суставов, формируются патологические изгибы позвоночника (кифоз поясничного отдела).
К концу первого года жизни лицо ребенка приобретает характерные черты по типу «гаргоилизма» – выступающие лобные бугры, широкая приплюснутая переносица, глазной гипертелоризм, толстые губы и язык, зауженная лицевая часть черепа. Типична задержка роста, который практически полностью останавливается к 4-5 годам. По мере прогрессирования заболевания присоединяются симптомы поражения центральной и периферической нервной системы.
Наблюдается заметное отставание психомоторного развития – интеллект снижен, речь неразвита. Дефектам речи также способствует формирующаяся нейросенсорная тугоухость. Из поведенческих расстройств отмечаются замкнутость и агрессия. Походка нарушена, тонус мышц снижен. Иногда возникают судороги вплоть до тонико-клонических пароксизмов.
Из других признаков синдрома Гурлера можно выделить частые инфекционные заболевания верхних дыхательных путей, рецидивирующие средние отиты, помутнение роговицы. Вследствие накопления ГАГ в миндалинах, трахее и надгортаннике постепенно сужается просвет дыхательной трубки, что повышает риск появления обструктивного апноэ сна.
Осложнения
Синдром Гурлера является тяжелым заболеванием с большим количеством осложнений. Основными причинами смерти считаются прогрессирующая хроническая сердечная недостаточность как исход формирующегося порока сердца и кардиомиопатии, дыхательная недостаточность из-за обструкции дыхательных путей, тяжелые инфекции (пневмония, менингит, туберкулез).
Гидроцефалия может привести к отеку головного мозга. Нарушения ритма сердца возникают вследствие фиброзного поражения миокарда. При компрессии спинного мозга возможны тетраплегия или нижняя параплегия, ухудшение или полная утрата тазовых функций. Иногда наблюдается потеря зрения и слуха.
Диагностика
Ввиду мультиорганного поражения пациентам с синдромом Гурлера необходим многопрофильный подход, поэтому курацию осуществляют врачи различных специальностей – педиатры, неврологи, кардиохирурги. Заподозрить наличие патологии помогают анамнестические данные и яркие фенотипические черты заболевания. Для подтверждения диагноза назначается дополнительное обследование, включающее:
- Определение ферментативной активности. В лейкоцитах периферической крови отмечается сниженная активность лизосомной α-L-идуронидазы.
- Экскреция ГАГ с мочой. Обнаруживается увеличение концентрации в моче гликозаминогликанов за счет дерматансульфата и гепарансульфата.
- МРТ головного и спинного мозга. На МРТ головного мозга просматривается утолщение мозговых оболочек, признаки гидроцефалии (расширение желудочков , цистерн), на спинальной МРТ– компрессия спинного мозга.
- Рентгенография. Рентгенографические признаки синдрома Гурлера включают расширение диафизов трубчатых костей, укорочение и утолщение ключиц, «веслообразную» форму ребер. На рентгенографии позвоночника отмечается его искривление, расширение позвонков, недоразвитие, деформация поперечных отростков.
- УЗИ сердца. Эхокардиография показывает утолщение или деформацию створок клапанов, признаки регургитации, расширение полостей сердца. При развитии ХСН снижается фракция выброса левого желудочка.
- УЗИ и КТ. При проведении УЗИ или КТ органов брюшной полости определяется диффузное увеличение печени и селезенки.
- Спирометрия. При измерении функции внешнего дыхания выявляются обструктивные нарушения – снижение форсированной жизненной емкости легких, максимального инспираторного потока.
- ЭНМГ. У некоторых пациентов при сдавлении нервных стволов на электронейромиографии обнаруживаются признаки нейропатии – блок и замедление проведения нервного импульса.
- Аудиометрия. Проведение аудиометрии показывает значительное увеличение порога звуковосприятия. Ввиду раннего возраста пациентов предпочтительны объективные методы аудиометрии (компьютерная).
- Офтальмоскопия. Очень часто при осмотре глазного дна определяется отек и застой диска зрительного нерва, периферическая дистрофия сетчатки.
- ДНК-диагностика. Решающим в диагностике заболевания считается молекулярно-генетическое исследование для выявления мутаций в гене IDUA.
Дифференциальный диагноз синдрома Гурлера проводится с другими наследственными метаболическими расстройствами – II, III типами мукополисахаридоза, ганглиозидозом, множественной сульфатазной недостаточностью. Поражение суставов следует отличать от неинфекционных артритов, ювенильного ревматоидного артрита.
Лечение синдрома Гурлера
Консервативная терапия
Пациенты подлежат обязательной госпитализации в стационар. Основным лекарственным патогенетическим лечением является пожизненная ферментная заместительная терапия (ФЗТ). Применяется рекомбинантная форма человеческой альфа-идуронидазы (ларонидаза). Введение этого препарата способствует восстановлению энзиматической активности, достаточной для гидролиза накопленных ГАГ и предотвращения их дальнейшего отложения.
Раннее использование ФЗТ позволяет замедлить прогрессирование неврологических расстройств и сердечной недостаточности, восстановить активные движения в суставах и позвоночнике, добиться регресса гепатоспленомегалии, исчезновения ночного апноэ. Также осуществляется следующая симптоматическая терапия:
- Кардиопрепараты. Для лечения застойной сердечной недостаточности назначаются ингибиторы АПФ (периндоприл), блокаторы бета-адренергических рецепторов (бисопролол), антагонисты альдостерона (спиронолактон).
- Антиконвульсанты. Для предотвращения возникновения приступов применяют противосудорожные медикаменты – антагонисты NMDA-рецепторов (фенитоин), стимуляторы активности ГАМК (габапентин), регуляторы ионных каналов.
- Психотропные средства. С целью коррекции поведенческих расстройств совместно с психоневрологом индивидуально подбирают транквилизаторы, седативные средства (бензодиазепины).
Хирургическое лечение
Больным синдромом Гурлера в возрасте до 2 лет рекомендовано радикальное лечение, позволяющее избежать необходимости в пожизненной ферментотерапии, – трансплантация гемопоэтических стволовых клеток (ТГСК). Проводится пересадка костного мозга либо стволовых клеток пуповинной крови HLA-совместимых родственных доноров. ТГСК предотвращает развитие нарушений когнитивных функций. Предварительно назначается ФЗТ и иммуносупрессивная терапия.
При наличии соответствующих показаний выполняются следующие виды операций:
- Протезирование клапана сердца: порок сердца (недостаточность или стеноз), приводящий к значимым гемодинамическим нарушениям и прогрессированию ХСН.
- Декомпрессия спинного мозга и нервных стволов: сдавление спинного мозга, вызывающее сенсомоторные нарушения и тазовые расстройства.
- Вентрикулоперитонеальное шунтирование: гидроцефалия с высоким давлением цереброспинальной жидкости.
- Грыжесечение: появление паховых или пупочных грыж (при ущемлении грыжи необходима экстренная операция).
- Тонзиллэктомия: выраженная гипертрофия миндалин, затрудняющая дыхание.
- Протезирование коленного или тазобедренного суставов: развитие грубых деформаций и контрактур, полностью ограничивающих активные движения.
Реабилитация
Важной частью лечения являются реабилитационные мероприятия, включающие два основных аспекта. Массаж и лечебная физкультура необходимы для восстановления движений в суставах. Психолого-педагогическая помощь в виде систематических индивидуальных занятий направлена на развитие когнитивных функций, способствует более длительному сохранению интеллекта.
Прогноз и профилактика
Синдром Гурлера – тяжелое инвалидизирующее заболевание с высоким процентом летальности. Средняя продолжительность жизни больных без своевременно назначенного патогенетического лечения составляет 10 лет. Наиболее частыми причинами смерти выступают дыхательная и сердечная недостаточность, тяжелые бактериальные инфекции.
Единственной эффективной профилактикой развития патологии является прерывание беременности, если во время пренатальной диагностики была обнаружена низкая активность альфа-идуронидазы при исследовании ворсин хориона на 8-10 неделе беременности. При наличии близкого родственника, который страдает синдромом Гурлера, рекомендуется проведение ДНК-диагностики. Для предотвращения инфекционных осложнений назначается вакцинация от пневмококка, менингококка, гемофильной палочки.
Литература
1. Болезни накопления, сопровождающиеся нарушениями нервно-психического развития. Наследственные нарушения нервно-психического развития у детей. Руководство для врачей/ под ред. Темина П.А., Казанцевой Л.З. – 2001.
2. Федеральные клинические рекомендации по оказанию медицинской помощи детям с мукополисахаридозом I типа — 2013.
3. Мукополисахаридоз I типа у детей. Клинические рекомендации — 2016.
4. Нарушения процессов клеточной биоэнергетики и методы их терапевтической коррекции у детей с мукополисахаридозами/ Семячкина А.Н., Сухоруков В.С.// Российский вестник перинатологии и педиатрии – 2005 — №1.
Мукополисахаридоз: короткая жизнь с надеждой на будущее

В этом году исполняется ровно 100 лет с тех пор, как канадец Чарльз Хантер составил первое описание мукополисахаридоза, выявленного у двух братьев. Сегодня известно, что это не одно, а целая группа заболеваний, очень редких. В мире проживает примерно 1,5 тысячи таких пациентов, около 200 из них живут в России. MedAboutMe выяснял, что такое мукополисахаридоз и какова судьбы людей, больных этим заболеванием.

Какие проблемы с кожей помогут решить средства с гиалуроновой кислотой?
Сухость кожи, чувство стянутости — всё это и другие проблемы, которые решают средства с гиалуроновой кислотой.
Мукополисахаридоз: ферменты и болезнь
Немалая часть нашего организма состоит из соединительной ткани. Ее клетки участвуют в формировании каркаса и наружного покрова всех органов тела. В некоторых их них соединительная ткань составляет до 90% от их массы. Это хрящи, кости, жир, фасции, синовиальная жидкость, которая плещется в суставах, лимфа, склера и радужка глаза, микроглия, окружающая нейроны и др.
Без преувеличения, главным элементом соединительной ткани можно считать протеогликаны. Это соединения, которые содержат белковое ядро и связанные с ним многочисленные и разнообразные гликозаминогликаны (ГАГ). В разных тканях — разные протеогликаны. Они образуются, живут 7-10 дней и распадаются под действием ферментов — лизосомных гидролаз. Последние расщепляют именно ГАГ, причем каждому виду гликозаминогликанов соответствует свой личный фермент. Процесс образования (анаболизма) протеогликанов и распада (катаболизма) идет постоянно.
Что произойдет, если нужного фермента не окажется? Нерасщепленные или частично разрушенные протеогликаны станут накапливаться, откладываясь в соединительных тканях, пронизывающих все наше тело. Так и развиваются мупоколисахаридозы — разновидность лизосомных болезней накопления.
Какие бывают мукополисахаридозы?

На сегодняшний день известно о 8 типах заболевания, причем некоторые из них представлены в нескольких разновидностях. Обычно это связано с тем, что дефицит одного фермента может быть вызван разными мутациями — и, как следствие, проявления этих мутаций и срок жизни больных тоже могут различаться.
- I тип объединяет три фенотипа: синдром Гурлер (МПС-IH), синдром Шейе (МПС-IS) и синдром Гурлер-Шейе (МПС-IH/S). Мутации, вызывающие синдром Гурлер и синдром Шейе, происходят в одном и том же гене. И иногда они развиваются одновременно. МПС I типа считается самой распространенной разновидностью болезни. Продолжительность жизни — от 6-10 (синдром Гурлер) до 30 лет (синдром Шейе).
- II тип — синдром Хантера (МПС-II). Это второй по частоте встречаемости вид мукополисахаридозов. Продолжительность жизни — 30-40 лет.
- III тип — синдром Санфилиппо, который подразделяется на 4 формы: A (МПС-III A), B (МПС-III B), C (МПС-III C) и D (МПС-III D). Это тоже разные мутации гена, который кодирует данный лизосомный фермент. Продолжительность жизни — до 20 лет.
- IV тип — синдром Моркио. Представлен двумя формами: A (МПС-IV A) и B (МПС-IV B). Продолжительность жизни — 20-35 лет.
- VI тип — синдром Марото-Лами (МПС-VI). Продолжительность жизни — 10-20 лет.
- VII тип — синдром Слая (МПС-VII).
- VIII тип — синдром Ди Ферранте (МПС-VIII) .
- XI тип — синдром дефицита гиалуронидазы, он же — синдром Натовича (МПС-IX).
Пятым типом раньше считался синдром Шейе.
Диагноз обычно ставят в детском возрасте.
МПС и генетика

Мукополисахаридоз — это генетическое заболевание. Оно наследуется по аутосомно-рецессивному типу. Это значит, что мутация только на одной хромосомы из пары (а мы помним, что у человека 23 пары хромосом) не вызовет заболевания, потому что ген, расположенный на парной нормальной хромосоме будет исправно работать. Благодаря этому организм будет производить нужный фермент в требуемых количествах. А вот если человек получит на обеих хромосомах одинаковые мутации в области генов, кодирующих один и тот же лизосомный фермент — у него разовьется мукополисахаридоз. Чтобы такая ситуация сложилась, оба его родителя должны быть носителями бракованного гена. Такое совпадение случается очень редко — и поэтому мукополисахаридозы относятся к орфанным (редким) заболеваниям.
Из общего списка выбивается синдром Хантера (или Гунтера, в зависимости от перевода), МПС II типа. Именно это заболевание стало первым из открытых человечеством мукополисахаридозов. В отличие от генов, являющихся причиной других видов МПС, ген, вызывающий синдром Хантера, расположен на половой X-хромосоме. То есть его наследование сцеплено с полом, и болеют им в подавляющем большинстве случаев мальчики.
- При других видах МПС, если болен один родитель, а второй здоров, ребёнок получит одну хромосому с мутантным геном, а другую — такую же, но нормальную. Болезнь не разовьется.
- Но половые хромосомы Х и Y отличаются друг от друга. Мальчики получают Y-хромосому от отца и X-хромосому от матери. Если мать была обладателем хотя бы одной Х-хромосомы с мутантным геном (то есть даже сама не болела, была лишь носителем), и эта «неправильная» хромосома вошла в геном ребёнка, то второй здоровой хромосомы ей в пару нет. Такой мальчик заболевает.
«Люди-горгульи» и другие проявления болезни
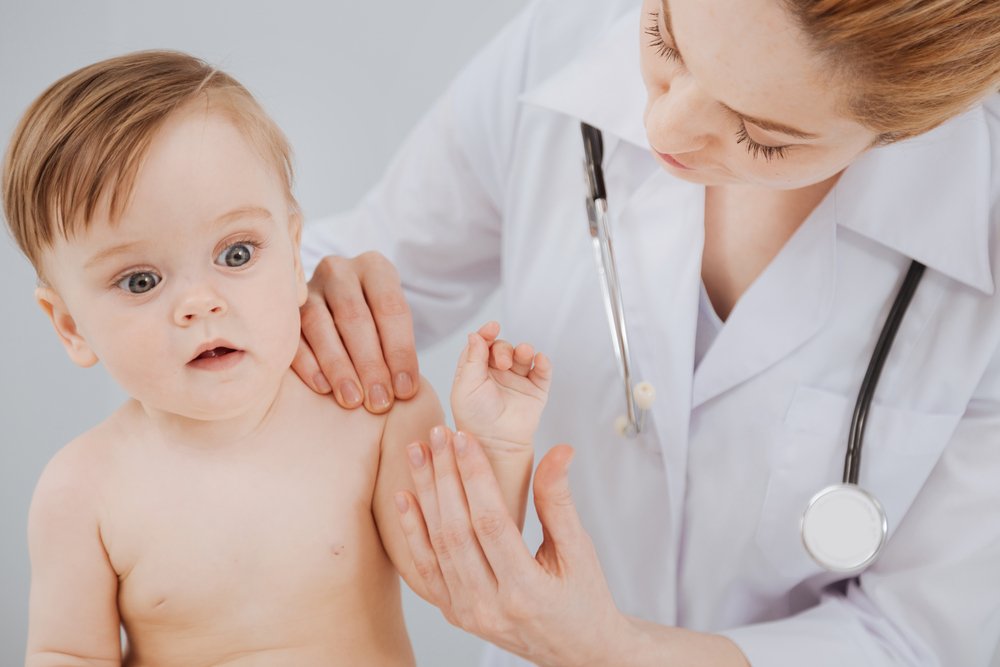
Разные типы мукополисахаридоза имеют свои характерные особенности. Например, при МПС I, II и III типов развиваются тяжелые нарушения нервной системы, которые приводят к деменции (слабоумию). А есть и общие черты. Как мы помним, речь идет о заболеваниях, связанных с соединительной тканью. Поэтому неудивительно, что на их фоне развиваются тяжелые системные поражения костей, суставов, кожи.
При этом может меняться форма головы, а при некоторых типах МПС уже с детства формируется специфическая внешность: приплюснутая переносица и нос с крупными ноздрями, широко посаженные глаза, толстые губы и приоткрытый рот, в целом грубые выразительные черты лица. Подметив сходство лиц таких пациентов с горгульями — каменными изображениями мифических существ характерного облика, врачи дали название данному состоянию — гаргоилизм (от французского слова gargouille).
Сегодня выделяют два основных фенотипа (внешние проявления) мукополисахаридоза:
- Гурлер-подобный фенотип. В этом случае обычно развиваются деменция, гепатоспленомегалия (увеличение печени и селезенки) и гаргоилизм.
- Моркио-подобный фенотип. У таких пациентов нормальный интеллект и не настолько выражены повреждения скелета и связок, как при Гурлер-подобном фенотипе.
Все пациенты с МПС также страдают от задержки роста (нанизм) и диспропорционального развития скелета, нарушений зрения (помутнение роговицы и др.), слуха (тугоухость), заболеваний сердца и сосудов (аритмии, гипертрофия миокарда, заболевания клапанов сердца), болезней дыхательной системы.
Лечение мукополисахаридозов
Специфическая терапия

Специфической называется терапия, мишенью которой являются непосредственные причины болезни. В данном случае это ферменты, которые не вырабатываются в достаточном количестве, и те нерасщепленные мукополисахариды, которые накапливаются и вызывают болезнь.
Только в середине прошлого века стали появляться препараты, позволяющие заменить недостающие ферменты. Сегодня производятся — и зарегистрированы в России — синтетические ферменты для трех типов МПС:
- МПС-I — Альдуразим (ларонидаза);
- МПС-II — Элапраза (идурсульфаза);
- МПС-VI — Наглазим (галсульфаза).
Недостаток Элапразы заключается в том, что она не проникает сквозь гематоэнцефалический барьер, то есть не попадает в мозг. В 2015 году СМИ сообщали о начале лечения 31-летнего пациента с синдромом Хантера новым препаратом AGT-182 — комбинацией идурсульфазы и специально разработанного для этого антитела. Предполагалось, что это позволит лекарству проникнуть в мозг. Результаты эксперимента пока не опубликованы.
Специфических лекарств для лечения других типов МПС в России пока нет. Только симптоматическая терапия, направленная на уменьшение проявлений болезни, дает возможность таким больным прожить отведенный им срок.
В 2014 году FDA одобрило препарат Вимизим (элосульфаза альфа), предназначенный для лечения МПС-IV А — разновидности синдрома Моркио. В России данный препарат не зарегистрирован.
Другой путь — субстратредуцирующая терапия, то есть угнетение выработки мукополисахаридов, избыток которых приводит к болезни. Пока такое лекарство разработано для МПС I, II и III типов (генистеин), но исследования проводились только на животных. В июне этого года ученые из Университета Манчестера сообщили о запуске проекта, в котором участвуют дети, больные МПС. Проект будет длиться год, и исследователи рассчитывают, что им удастся остановить разрушение центральной нервной системы при этом заболевании.
Трансплантация костного мозга
Ферментозаместительная терапия основана на введении экзогенных (то есть извне) синтетических ферментов. А трансплантация костного мозга (ТКМ) призвана заставить организм вырабатывать собственные, эндогенные ферменты при помощи пересаженных донорских стволовых клеток.
ТКМ — крайне сложная и очень дорогостоящая процедура. Она подходит далеко не всем пациентам и не со всеми типами МПС. Лучшие результаты были получены при пересадке костного мозга детям, страдающим МПС I типа, в возрасте 2 лет, то есть до того момента, пока поражение центральной нервной системы не стало слишком обширным. При попытках лечения методом ТКМ мукополисахаридозов II типа отмечается повышенная частота осложнений и летальность среди пациентов. Следует учитывать, что и сам процесс подбора неродственного донора возможен далеко не для всех пациентов.
Генная терапия

Одним из самых значимых направлений в области разработки методов лечения МПС является генная терапия при помощи вирусных векторов — кольцевых ДНК, способных переносить нужный ген и встраивать его в заданный участок хромосомы. И уже есть первые положительные результаты.
В июле этого года ученые из Университета Миннесоты рассказали, как закапали в нос экспериментальным мышам препарат, содержащий вирус с геном, кодирующим лизосомный фермент альфа-L-идуронидазу — именно его так не хватает больным с МПС I типа. Введение препарата через нос позволило ему проникнуть через гематоэнцефалический барьер в мозг и защитить мозг от разрушительного действия накапливающихся мукополисахаридов. Теперь ученые ищут возможности повторить ту же самую процедуру с участием людей.
Мукополисахаридоз в России
С 2012 года мукополисахаридоз входит в список жизнеугрожающих орфанных заболеваний и подлежит лечению по программе «7 нозологий». Если быть точными, то в список вошли только те типы МПС, от которых есть лекарства и при этом они зарегистрированы в нашей стране, то есть: МПС-I, МПС-II и МПС-VI.
Сегодня в России, по данным общественных организаций, занимающихся проблемой МПС, проживает около 200 пациентов с мукополисахаридозом. А по данным компании Санофи, в 2017 году диагноз МПС имели 98 россиян, причем 88 из них — это дети. Сегодня лечение получают только 77 из них: 52 человека находятся на пожизненной ферментозаместительной терапии, а 25 — прошли трансплантацию костного мозга (при синдроме Гурлер эта операция может проводиться только в возрасте от 2 до 4 лет). Из-за высокой стоимости лекарств, которые следует вводить еженедельно, расходы на это заболевание лидируют в бюджетах, выделенных на лечение орфанных болезней.
Например, для лечения людей с синдромом Хантера необходим препарат Элапраза. Стоимость одного флакона колеблется от 100 до 200 тысяч рублей. Для лечения одного пациента еженедельно требуется 300-500 тысяч рублей. И это пожизненная терапия. Должна быть.
В последние годы закупкой препаратов для пациентов с МПС занимались региональные власти. Нежелание тратить огромные деньги на отдельных пациентов с редкими болезнями приводило к бесконечным задержкам поставки лекарств — а это немедленно ухудшало состояние больных. Но недавно, в июне стало известно, что финансирование закупок препаратов для лечения МПС с будущего, 2018 года будет производиться из федерального бюджета. Это дает надежду многим из нынешних пациентов дожить до того момента, когда их болезнь можно будет вылечить.
Читайте далее

Какие болезни диагностируют по слюне?
Как проводят диагностику по слюне, какие болезни можно так обнаружить, и почему это лучше анализа крови — в статье MedAboutMe
Какие проблемы с кожей помогут решить средства с гиалуроновой кислотой?
Сухость кожи, чувство стянутости — всё это и другие проблемы, которые решают средства с гиалуроновой кислотой.
Генетические заболевания — лечение в Германии

Лечение генетических заболеваний в Германии ограничивается симптоматическим лечением. Гораздо важнее — генетическая диагностика на этапе планирования беременности, осознание рисков, определение методов профилактики.
Генетические или, как их еще называют, наследственные болезни могут быть вызваны мутацией в определённом гене или же несколькими мутациями в нескольких генах. Поэтому среди генетических заболеваний выделяют моногенные и полигенные.
Наследственные заболевания всё же отличаются от генетических, так как они вызваны нетипичными изменениями в гене и передаются из поколения в поколение. Самые яркие примеры — гемофилия или дальтонизм.
Генетические заболевания, помимо наследственных, включают также спонтанные изменения в геноме человека. Как, например, при трисомии (это наличие трёх гомологичных хромосом вместо пары, самым известным примером трисомии является болезнь Дауна, которую часто называют трисомией по хромосоме 21). Изменения в хромосомном составе происходят самопроизвольно во время деления клеток эмбриона и не наследуются от родителей.
Наследственные заболевания имеют различную степень вероятности наследования. Различают аутосомно-рецессивный и аутосомно-доминантный тип наследования. Болезни могут быть связаны как с нарушением ядерной, так и митохондриальной ДНК.
Аутосомно-рецессивный тип наследования
Заболевание проявляется только в том случае, если изменения определённого гена есть в каждой из двух хромосом, когда мутированный ген наследуется и от биологического отца, и от матери. Родители при этом не всегда имеют признаки болезни, а являются только носителями изменённого гена. Примерами аутосомно-рецессивных типов наследования являются муковисцидоз, альбинизм и фенилкетонурия.
Заболевания по аутосомно-рецессивному типу наследования
Врожденная гиперплазия надпочечников (АГС);
- Лейциноз;
- Альбинизм;
- Алкаптонурия;
- Дефицит Alpha1-антитрипсина;
- Галактоземия;
- Наследственная непереносимость фруктозы;
- Синдром Жубера;
- Кретинизм;
- Полидактилия;
- Синдром Барде-Бидля;
- Расщепление губы и нёба (заячья губа, волчья пасть);
- Болезнь Вильсона-Коновалова;
- Мукополисахаридоз (МПС);
- Муковисцидоз (кистозный фиброз);
- Нефротический синдром;
- Аномалия Петерса;
- Фенилкетонурия;
- Синдром Ангельмана;
- Талассемия;
- Ксеродерма;
- Поликистоз почек.
Аутосомно-доминантный тип наследования
Генетическая болезнь развивается вследствие наличия в гене изменённой аллели в одной из двух гомологичных хромосом. Генетическая информация с патологическим отклонением закладывается на одной из 44 аутосом (неполовые хромосомы) и наследуется независимо от пола, если у человека есть хотя бы один изменённый аллель в аутосоме. Случаи заболевания диагностируются в каждом поколении и наследуются вертикально по родословной.
Заболевания по аутосомно-доминантному типу наследования
- Болезнь Гентингтона;
- Ретинобластома;
- Полидактилия;
- Парциальный альбинизм;
- Ахондроплазия;
- Синдром Аперта;
- Брахидактилия;
- Синдром Элерса — Данлоса;
- Синдром Ангельмана;
- Протопорферия;
- Эритропоэтическая протопорфирия;
- Фактор 5, мутация Лейдена;
- Гиперлипидемия;
- Наследственная моторно-сенсорная нейропатия (Шарко-Мари-Тута);
- Злокачественная гипертермия;
- Синдром Марфана;
- Миотоническая дистрофия;
- Нейрофиброматоз;
- Несовершенный остеогенез;
- Синдром Банаян-Райли-Рувальбака;
- Серповидноклеточная анемия;
- Поликистоз почек.
Болезни, сцепленные с Х- или Y-хромосомами
Большинство генетических болезней связаны с мутацией в Х-хромосоме, так как Y-хромосома содержит меньше генов.
X-сцеплённое рецессивное наследование
У женщин болезнь проявляется только в случае, если обе Х-хромосомы повреждены, в иных случаях они являются только носителями и передают их далее своим детям. Женщины могут компенсировать патологические изменения в Х-хромосоме благодаря наличию второй Х-хромосомы, если она не изменена. У мужчин заболевание проявляется, если они получают унаследованную измененную Х-хромосому от фенотипически здоровой матери, или одну из двух патологически изменённых Х-хромосом фенотипически больной матери, потому что мужчины на самом деле имеют только одну Х-хромосому в генотипе и наследуют её от матери.
Примерами являются:
- Дефицит глюкозо-6-фосфат-дегидрогеназы;
- Гемофилия А и В;
- Синдром Лёша — Нихена;
- Болезнь Фабри;
- Мукополисахаридоз II типа;
- Мышечная дистрофия (типа Дюшенна, Беккера Кинер);
- Болезнь Норри;
- Пигментный ретинит;
- Дейтеранопия;
- Септический гранулематоз;
- Тяжёлый комбинированный иммунодефицит (X-SCID);
- Дефицит орнитин-карбамоилтрансферазы (OTC).
X-сцеплённое доминантное наследование
50% мужчин имеют проявления заболевания в случае, ели их мать — носитель патологически изменённой Х-хромосомы. Если же мать имеет обе патологическе Х-хромосомы, то все дети будут затронуты. У женщин вероятность проявления болезни выше, т.к. они имеют 2 Х-хромосомы (одна от матери, одна от отца), в то время как у мужчин всего одна Х-хромосома.
Примерами могут служить:
- Синдром де Тони — Дебре — Фанкони (первичный изолированный синдром Фанкони, глюкозо-фосфат-аминовый диабет);
- Синдром Ретта.
Митохондриальные и внехромосомные наследственные болезни
Около 0,1 процента ДНК человеческой клетки располагается не в ядре, а в митохондриях. Так как яйцеклетки, в отличие от сперматозоидов имеют сотни тысяч митохондрий, мутации в митохондриальной ДНК наследуются преимущественно только от матери. Помимо довольно распространённой митохондриальной миопатии, среди этого типа болезней выделяются также митохондриальный сахарный диабет, наследственная оптическая нейропатия Лебера, синдром Вольфа-Паркинсона-Уайта, рассеянный склероз, синдром Лея, митохондриальная нейрогастроинтенстинальная энцефалопатия.
Диагностика и лечение наследственных и генетических заболеваний в Германии
При существовании подозрения возможной наследственной болезни, назначается генетическое исследование. Хромосомы проверяются на наличие количественных и структурных изменений. Если подозрение конкретного генетического дефекта будет подтверждено в ходе такого предварительного обследования, возможно назначение также более широкого и комплексного изучения структуры гена. Результаты могут быть полезны при определении рисков появления определенной наследственной патологии на этапе планирования беременности или в ходе пренатальной диагностики и определении эффективного метода профилактики.
Основные методы диагностики: клинико-генеалогический, цитогенетический, биохимический, иммунологический, молекулярно-генетический (ДНК-анализ), методы пренатальной диагностики.
Терапевтически невозможно воздействовать на генетически унаследованную особенность человека. Возможно, только дать совет относительно дальнейшего образа жизни, осознавая все возможные риски, а также принимать симптоматические меры лечения. В большинстве случаев речь идет не о болезни, а о восприимчивости человека к определённым факторам и изменении образа жизни, как части комплексной терапии болезни. Главной остаётся профилактика наследственных патологий посредством диагностики и определения рисков.
Поделиться
Добавить комментарий
Авторизация при помощи социальных сетей
Комментарии
0 Арби 30.04.2019
Здравствуйте. Читал почти все комментарии о Ретт Синдроме. У дочери такой диагноз ,хотелось бы узнать на сегодняшний день есть ли какое нибудь лекарство или где находится в Европе лучший реабилитационный центр по этому диагнозу. Спасибо заранее. Всем здоровья. Благодарю за информацию.
0 Сергей Гринчук 04.05.2019
Здравствуйте, на сегодняшний день для синдрома Ретта предполагается только поддерживающие методы лечения, которые могут благоприятно повлиять на некоторые области и предотвратить инвалидность. Не все методы реабилитации представленные ниже, подходят для любого ребенка с этим синдромом. Выбор сочетания этих методов — задача врача, но стоит уделить внимание их комбинированию: физиотерапия, иппотерапия, кетогенная диета, музыкотерапия, логопедия, водолечение, Доман-терапия, терапия по Петё. Устранить причину болезни, мутацию в гене, современной медицине пока не под силу. Из реабилитационных центров стоит упомянуть Клинику Фогтаройт, Центр социальной педиатрии при Клинике Гросхадерн, отделение нейрологии Клиники Швабинг.
0 Иван Иванов 16.04.2019
Здравствуйте у врачей есть подозрения что у ребенка синдром меласа (1.5 года) что делать?помогут ли где нибудь?
0 Сергей Гринчук 04.05.2019

На основе чего был поставлен диагноз? Если диагноз был поставлен верно, то, поскольку это наследственное заболевание, терапия причины, мутации гена, в настоящее время невозможна. В качестве общего метода лечения следует позаботиться о том, чтобы обеспечить достаточное снабжение пациента энергией в форме глюкозы и жиров, жидкостей и минералов, а также избегать условия повышенного потребления энергии. Это включает, например, заблаговременное снижение высокой температуры (жара), потому что с повышением температуры повышается метаболизм и потреблением энергии. Таким образом можно избежать судорог, связанных со значительно возросшим потреблением энергии. Следует позаботиться о том, чтобы избегать лекарств, которые подавляют дыхательную цепь. Среди них, например, противосудорожный вальпроат. Существуют попытки лечения витаминами и так называемыми кофакторами дыхательной цепи (коэнзим Q10, биотин, тиамин, креатин), но пока нет точных доказательств их эффективности. Среди клиник, в которых можно пройти диагностику, получить консультацию по организации дальнейшей жизни пациента и начать реабилитацию, следует отметить Центр лечения митохондриальных болезней при Клинике Швабинг, Центр врождённых болезней нарушения обмена веществ при Клинике Людвига-Максимилиана, Центр митохондриальных заболеваний при Клинике Людвига-Максимилиана.
Если же Вы не уверены, что у ребёнка именно этот диагноз, стоит приехать на полноценную диагностику в Мюнхен. Напишите, пожалуйста, на почту infomed-svet.de с указанием номера телефона для связи, мед. документацией о пройденных на данный момент обследованиях, можно на русском языке. Эта информация необходима для составления запроса в клинику для получения информации о стоимости диагностики.0 Холназаров Шариф 11.01.2019
Здравствуйте у моей дочки синдром Жубер ей 9 месяцев мы провели все необходимые анализы в Индии там определилась все все ответы есть на английском можно ли её лечить в Германии если да то на сколько процентов это возможно?
0 Сергей Гринчук 16.01.2019
Здравствуйте, Вы уверенны, что это синдром Жильбера? Почему проводилась диагностика? И какая именно? Генетическая? Анализ крови?
Вы можете прислать свои медицинские документы на английском языке.
Но синдром Жильбера не лечится. Это генетический дефект, который невозможно изменить. Просто нужно придерживаться определённых правил на протяжении жизни: соблюдать правильное и регулярное питание, избегать жажды, избегать нагрузки на печень и т.д.0 Лаура 15.12.2018
Спасибо за ответ. У меня еще один вапрос. Сейчас проводится ли эксперементалные операции по удалению мутираваннова гена и замены человека?
0 Сергей Гринчук 15.12.2018
Нет. То, что китайский учёный утверждает, что в Китае были рождены близнецы с заменой гена, во-первых не означает, что это правда, во-вторых проще заменить ген у зиготы в пробирке, чем у взрослого человека. Такой технологии не существует. Пока.
-1 Лаура 07.12.2018
у меня диагноз шарко-мари-тута . Я бы хотела узнат ест ли личение этой болезни в германии или проводится ли эксперементалные личение.
0 Сергей Гринчук 07.12.2018
Добрый день. На основании чего Вам был поставлен диагноз? Генетическое исследование? Измерение скорости проводимости нерва или биопсия нерва? Излечение генетического заболевания на данном этапе развития медицины не возможно. Лечение симптоматическое: физиотерапия и хирургическое вмешательство для сохранения функциональности нижних конечностей.
На данный момент проходит международная исследовательская работа, в которой также принимает участие Университетская клиника Людвига-Максимилиана. Исследование начато в 2015 году. Сейчас — уже 3 фаза наблюдений. Испытывается препарат, облегчающий симптомы. Но результаты еще не предают огласке.0 Анар 04.04.2018
нам поставили диагноз синдром Ретте нашей доченьке полтора годика хотели бы как нибудь пролечить
0 Сергей Гринчук 10.04.2018К сожалению, на сегодняшний день нет лечебной терапии синдрома Ретте. В клиниках Германии Вам могут предложить реабилитационную терапию: физиотерапию, эрготерапию, гидротерапию и т.д. Цель данной терапии — развитие, восстановление и поддержание навыков, необходимых ему для самообслуживания. Вашей доченьке сейчас 1,5 года. По немецкой классификации болезнь находится на 1 стадии: Стадия застоя (от 6 до 18 месяцев). Это лучшее время для начала терапии на раннем этапе. Кроме того этот синдром можно спутать с другими нарушениями в развитии на раннем этапе, например с аутизмом. Я советую Вам пройти диагностику. Вы делали генетический тест?
Если Вы заинтересованы в дальнейшей консультации, вышлите, пожалуйста, Ваши актуальные мед. документы на русском, английском или немецком на почту infomed-svet.de .Источник https://www.krasotaimedicina.ru/diseases/genetic/Hurler-syndrome
Источник https://medaboutme.ru/articles/mukopolisakharidoz_korotkaya_zhizn_s_nadezhdoy_na_budushchee/
Источник https://med-svet.de/genetika-v-germanii