Синдром Марфана
Синдром Марфана — дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии. У больных с синдромом Марфана отмечаются гигантизм, долихостеномелия и арахнодактилия, аневризмы аорты, миопия, эктопия хрусталика, деформация грудины, кифосколиоз, плоскостопие, протрузия вертлужной впадины, эктазия твердой мозговой оболочки. Диагноз синдрома Марфана основан на семейном анамнезе, результатах функционального, офтальмологического, рентгенологического и генетического исследований. Лечение при синдроме Марфана включает консервативную и хирургическую коррекцию сердечно-сосудистых нарушений, поражений скелета и органа зрения.
МКБ-10
Q87.4 Синдром Марфана
- Причины синдрома Марфана
- Классификация синдрома Марфана
- Симптомы синдрома Марфана
- Диагностика
- Лечение синдрома Марфана
- Прогноз
- Цены на лечение
Общие сведения
Синдром Марфана — системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. Синдром Марфана — одна из наиболее распространенных наследственных коллагенопатий синдромального характера. Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.
Синдром Марфана
Причины синдрома Марфана
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью. В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки. Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).
Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене FBN1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста — TGFBR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных — первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
Классификация синдрома Марфана
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
- стертую — со слабо выраженными изменениями в 1-2-х системах
- выраженную — со слабо выраженными изменениями в 3-х системах; выраженными изменениями хотя бы в 1-ой системе; выраженными изменениями в 2-3-х и более системах.
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
Симптомы синдрома Марфана
Синдром Марфана характеризуется сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем; многообразием проявлений, варьированием сроков появления первых признаков заболевания; хроническим прогредиентным течением.
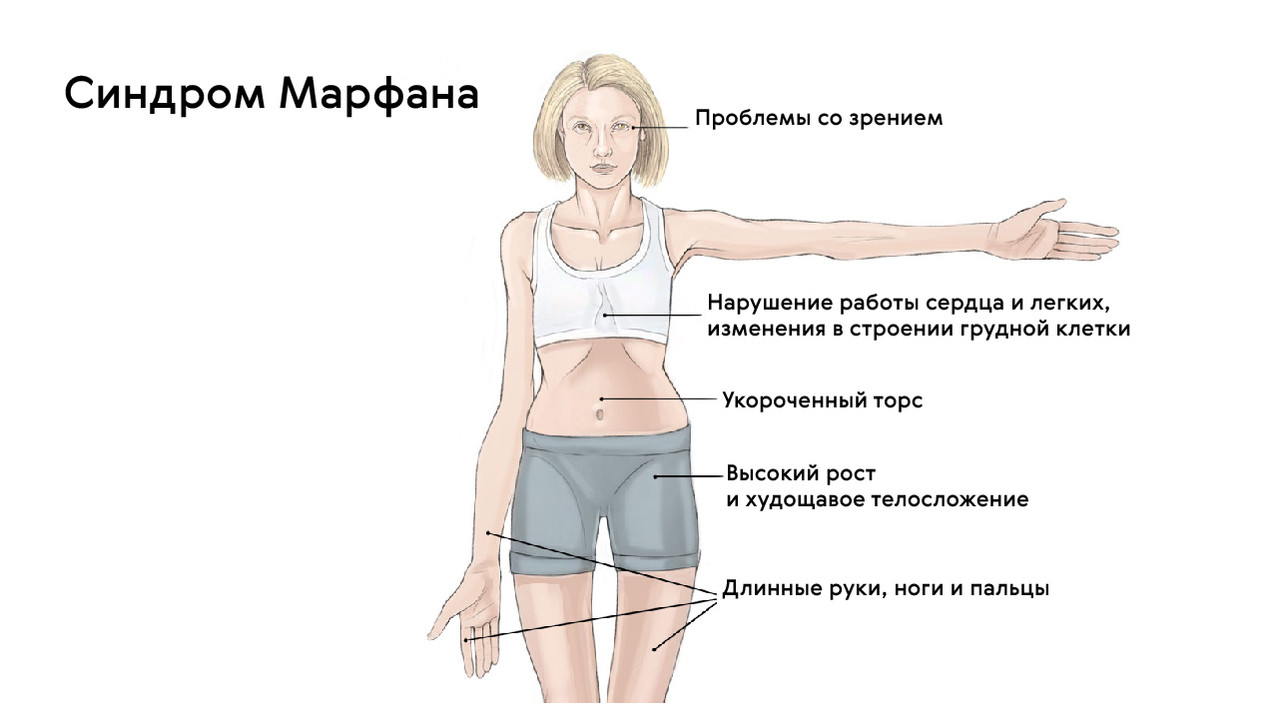
Больные синдромом Марфана, как правило, отличаются высоким ростом, относительно коротким туловищем с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными паукообразными пальцами (арахнодактилией); астеническим телосложением со слаборазвитой подкожной клетчаткой и мышечной гипотонией; длинным и узким лицевым скелетом (долихоцефалией); наличием высокого аркообразного неба и нарушения прикуса (прогнатии). Средняя длина тела при рождении у мальчиков с синдромом Марфана составляет 53 см, окончательный рост – 191 см; у девочек — соответственно 52,5 см и 175 см.
При синдроме Марфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия вертлужной впадины.
Сердечно-сосудистая патология, доминирующая в клинической картине синдрома Марфана и часто определяющая его исход, проявляется дефектами структуры стенок сосудов эластического типа, особенно аорты и крупных ветвей легочной артерии, пороками развития клапанного аппарата и перегородок сердца. Изменения аорты у больных синдромом Марфана характеризуются прогрессирующим расширением ее восходящей части и клапанного кольца (дилатацией, аннулоаортальной эктазией) и аневризмами; поражение митрального клапана — миксоматозной дегенерацией створок, патологическим удлинением и разрывом створочных хорд, обызвествлением клапанного кольца. У плода с синдромом Марфана возможно формирование врожденных пороков сердца — коарктации аорты, стеноза легочной артерии, ДМПП и ДМЖП. Органические и функциональные изменения сердца и сосудов у больных синдромом Марфана часто сопровождаются нарушением ритма (наджелудочковой и желудочковой тахикардией, фибрилляцией предсердий) и развитием инфекционного эндокардита.
Самая неблагоприятная неонатальная форма синдрома Марфана проявляется в классическом варианте уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни ребенка.
Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая близорукость, вывих/подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы, гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки. Эктопия хрусталика при синдроме Марфана имеет двухсторонний характер, часто развивается в возрасте до 4-х лет и устойчиво прогрессирует, ухудшая зрительную функцию.
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховые и бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и др.
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Диагностика
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему 1,05; сколиоз (> 20˚) или спондилолистез; ограничение разгибания в локтевом суставе (
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ — обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов — выявить дилатацию и аневризмы аорты.
Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Необходима дифференциальная диагностика с заболеваниями, внешне напоминающими синдром Марфана: гомоцистинурией, врожденной контрактурной арахнодактилией (синдромом Билса), наследственной артроофтальмопатией (синдромом Стиклера), MASS-синдромом, синдромами Элерса-Данлоса, Лойса-Дитца, Шпринцена–Голдберга, семейной эктопией хрусталика и др.
Лечение синдрома Марфана
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости. При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Синдром Марфана

Синдром Марфана — наследственное заболевание, которое проявляется системным поражением соединительной ткани в организме человека. В результате болезни происходят нарушения строения скелета и кожи, работы глаз, сердечно-сосудистой, дыхательной и других систем организма. Эту генетическую мутацию нельзя предотвратить или вылечить, но правильно подобранное лечение способно продлить пациентам жизнь и предупредить развитие опасных осложнений.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
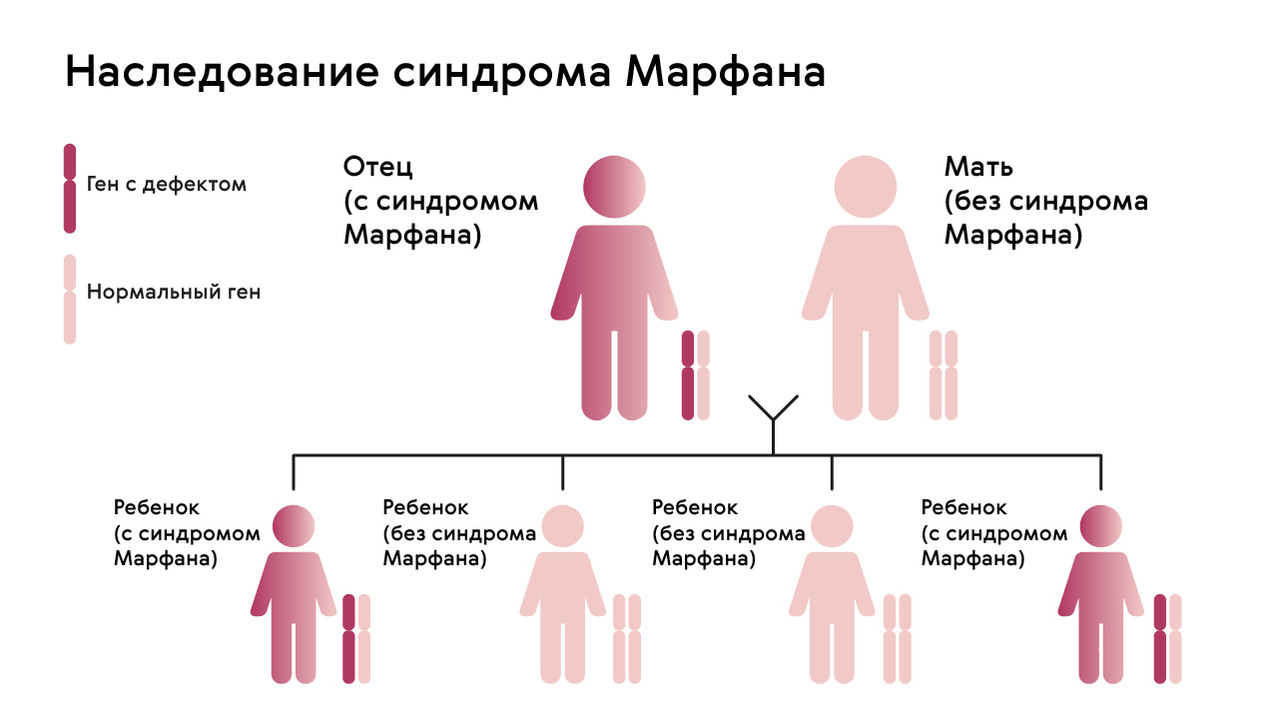
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на 5–10 тысяч.
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно. Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25–50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Симптомы синдрома Марфана
Проявления генетического дефекта могут быть выражены в разной степени: от легкого изменения строения соединительной ткани до тяжелых нарушений жизненно важных функций организма. Иными словами, внешние признаки аномалии у разных пациентов могут значительно отличаться, несмотря на одинаковый генетический дефект.
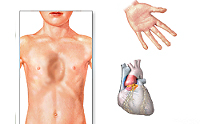
Классической триадой синдрома Марфана считаются: скелетные нарушения, смещение хрусталика и расслоение аорты (рис. 2). Также системное поражение соединительной ткани у пациентов становится причиной развития нарушений работы практически всех органов и систем организма.
Костно-мышечная система
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести случая и особенностей организма пациента.
Для людей с синдромом Марфана характерен чрезвычайно высокий рост: обычно дети «перерастают» всех членов семьи. При этом часто, особенно в детском возрасте, привлекает внимание нестандартная длина рук: их размах оказывается больше, чем длина тела.
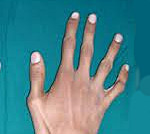
Яркий симптом болезни — патологически удлиненные и тонкие пальцы, так называемые «пальцы паука» (арахнодактилия) (рис. 3).
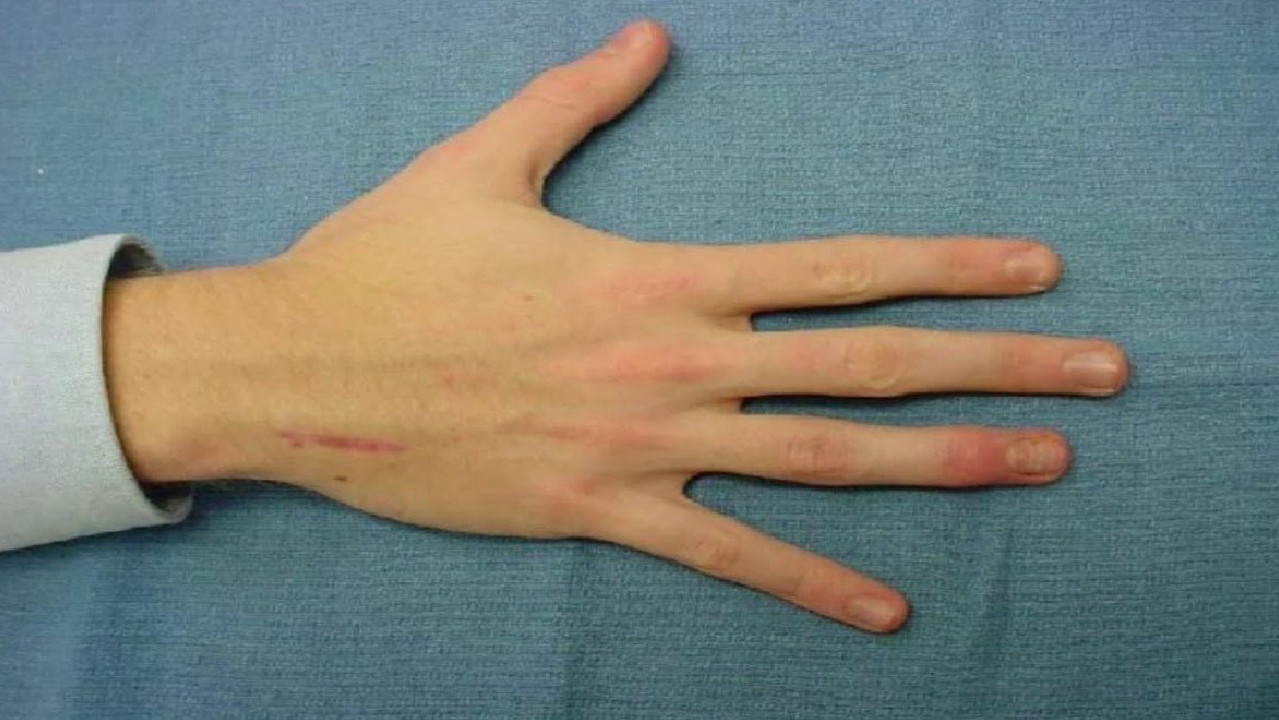
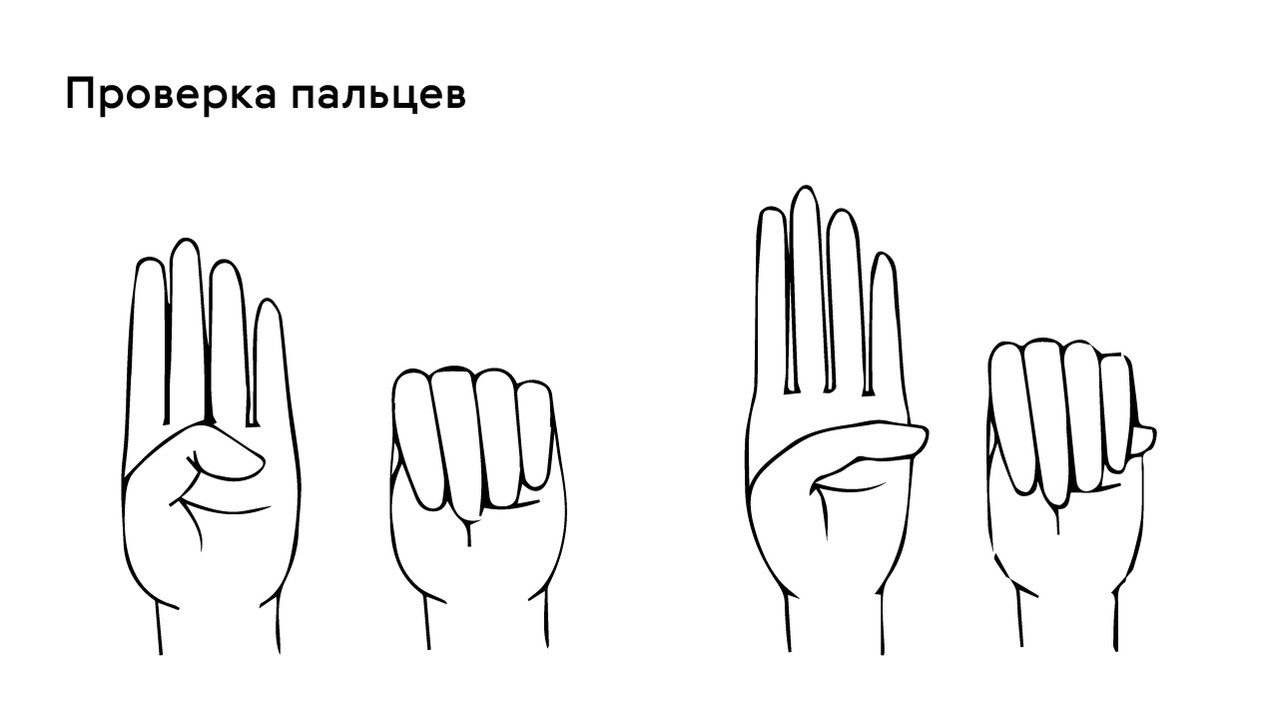
Проверить наличие симптома можно с помощью теста большого пальца кисти — у пациентов с арахнодактилией часть большого пальца (дистальная фаланга) выступает за край сжатой в кулак ладони (рис. 4).
Лицо людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
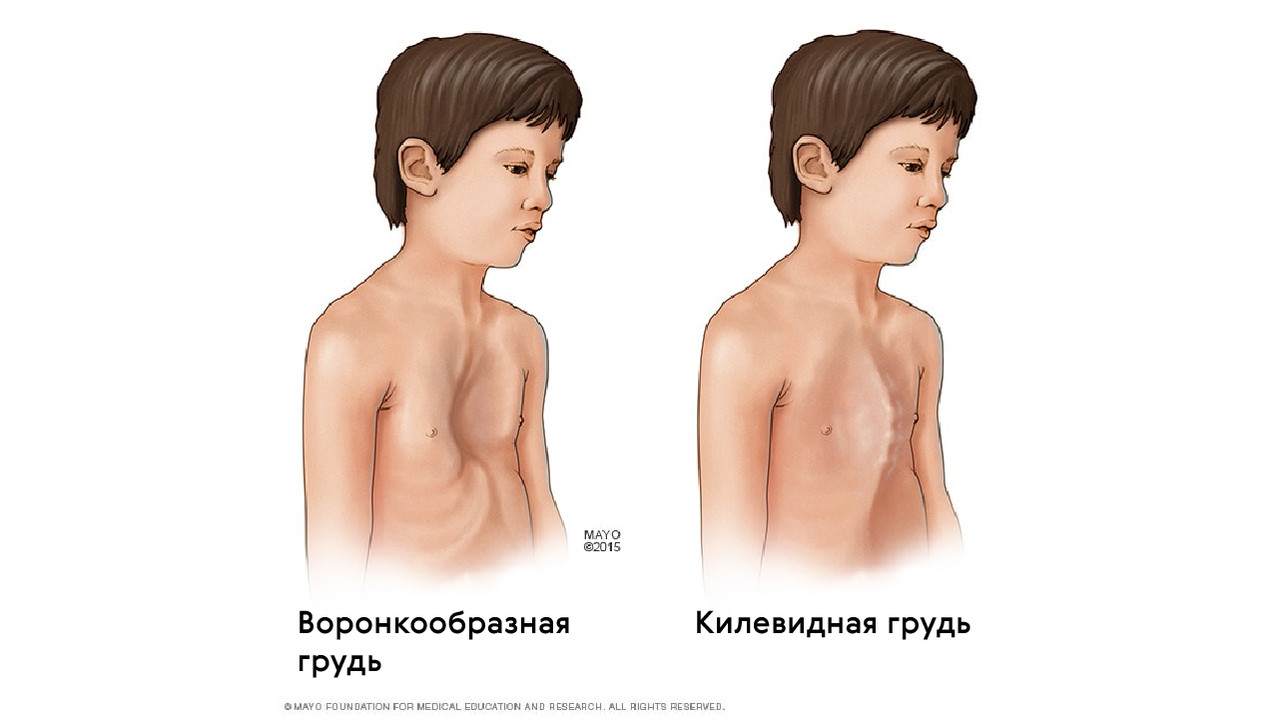
Также для таких пациентов характерны деформации грудной клетки, которые могут быть в двух вариантах: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь, рис. 5).
Осанка пациентов с синдромом Марфана в большинстве случаев нарушена. Чаще всего определяются различные степени выраженности сколиоза (отклонение позвоночного столба в сторону) или кифоза (формирование «горба»).
Кроме того, пациенты с FBN1 мутацией часто страдают от:
- плоскостопия;
- повышенной подвижности всех суставов;
- слабости связочного аппарата.
У пациентов с синдромом Марфана часто плохо развиты мышечные структуры и практически нет подкожно-жирового слоя. Движения пациентов с этой патологией неловкие, они часто получают различные травмы.
Кожа
Высокий темп роста и нарушения выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется в виде повышенной растяжимости кожных структур с образованием светлых полос — «растяжек» (стрий).
Зрение
Дефекты гена FBN1 определяют склонность к патологиям зрительной системы. Чаще всего повреждения глаз у пациентов с синдромом Марфана включают в себя:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Кроме того, у таких пациентов гораздо раньше может развиться катаракта или глаукома: те патологии органа зрения, которые считаются возрастными у здоровых людей.
Органы дыхания
В легких пациентов с синдромом Марфана может патологически разрастаться соединительная ткань. Это приводит к формированию сужения бронхов и легочного фиброза. Нередко на фоне генетической мутации развивается бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет возможность развития спонтанного пневмоторакса — неотложной ситуации, в которой в полость вокруг легких попадает воздух, и легкое резко уменьшается в размерах («спадается»).
Желудочно-кишечный тракт
Процессы пищеварения у людей с FBN1 мутацией меняются: нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз.
Почечный аппарат
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Нервная система и психическая сфера
Хотя в большинстве случаев у пациентов с синдромом Марфана не происходит нарушений работы мозговых структур, некоторые патологические изменения нервной системы могут присутствовать. Например, расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. Для таких пациентов характерно развитие синдрома хронической усталости — астения, склонность к депрессии. Интеллектуальная деятельность в большинстве случаев не нарушена, даже наоборот: среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Сердечно-сосудистая система
Кардиологи выявляют нарушения ритма сердца у людей с синдромом Марфана. У пациентов с этой патологией часто нарушается структура аортального клапана — соединительнотканной перегородки, которая предупреждает обратный ток крови из аорты в сердце. Это приводит к развитию порока сердца — аортальной недостаточности. Также могут развиваться другие пороки сердца, например, пролапс или недостаточность митрального клапана, а на пораженных участках часто развивается инфекционно-воспалительный процесс — бактериальный эндокардит.
Самую большую опасность представляют патологические изменения в главном сосуде организма — аорте. У 65–100% людей с синдромом Марфана есть большой риск поражения луковицы (наиболее близкая к сердцу часть аорты) и восходящей дуги этой артерии — тех частей, которые непосредственно выходят из сердца. Поскольку внутренний слой стенки сосудов также содержит волокна соединительной ткани, они склонны к износу, а давление крови в аорте выше, чем в других участках сосудистого русла. Это приводит к тому, что сосуд постепенно расширяется, и может произойти патологическое скопление крови между сосудистыми стенками с формированием мешковидного выпячивания (аневризмы) или спонтанный разрыв артерии.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка. При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).

Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7–18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка. Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны. Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития.
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает 70 лет. Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.
Заключение
Конечно, жизнь с этой генетической мутацией становится сложнее, но при правильном подходе к собственному здоровью и своевременному обследованию у врачей пациентам с синдромом Марфана удается компенсировать все проявления заболевания и не допустить развития фатальных осложнений.
Активисты с синдромом Марфана создают тематические сообщества по всему миру: мощная поддержка людей с такой же генетической аномалией позволяет пациентам не чувствовать себя одинокими.
Источники
- Отделение синдрома Марфана. Андалузский институт высокотехнологичной кардиологии (Бенальмадена и Фуэнхирола, Малага, Испания).
- Dadkhah, E., Ziaee, M., Davari, M. H., Kazemi, T., & Abbaszadegan, M. R. (2012). Informative STR Markers for Marfan Syndrome in Birjand, Iran. Iranian journal of basic medical sciences, 15(5), 1020–1025.
- Прийма, Н. Ф., Попов, В. В., Комолкин, И. А., & Афанасьев, А. П. (2013). Аневризма аорты у пациента с синдромом Марфана. Педиатр, 4 (1), 100-108.
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010 Jul;47(7):476-85. doi: 10.1136/jmg.2009.072785. PMID: 20591885.
Что такое Синдром Марфана: описание патологии и варианты лечения
Синдром Марфана – это наследственная аутосомно-доминантная патология соединительной ткани.

Общие данные
Синдром был открыт в 1875 году офтальмологом из Америки Э. Вильямсом. Он обнаружил полное смещение хрусталика глаза от своего нормального положения у брата и сестры, которые имели высокий рост и чрезмерную подвижность суставов с рождения. Спустя несколько лет детский врач из Франции А. Марфан вел наблюдения за девочкой 5 лет с прогрессирующими аномалиями скелета, чрезмерно длинными конечностями и «паучьими пальцами». Он дал четкое описание патологии, благодаря чему синдром получил его имя.
Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.
По данным американского Национального Фонда Синдрома Марфана, в США проживает около 200 000 человек с этим заболеванием, и лишь половина из них диагностированы. Представители фонда активно информируют общественность о признаках этого заболевания, чтобы люди могли обратиться за медицинской помощью, ведь это заболевание встречается гораздо чаще, чем многие думают.
Синдром Марфана развивается вследствие дефекта (изменения) в гене, который определяет структуру фибрина, который играет огромную роль в соединительной ткани. Человек с синдромом Марфана рожден с нарушением, даже если это не было установлено в течение жизни. Хотя каждый человек с синдромом Марфана имеет дефект определенного гена, изменения специфичны для каждой семьи и не каждый может испытывать подобные симптомы в одинаковой степени. Это называют подразумевает, что дефектный ген проявляется у различных людей в различной степени.
Дефектный ген может быть унаследован: ребенок имеет 50% вероятность унаследовать заболевание от родителя с синдромом Марфана. Иногда новый генный деффект появляется во время соединения сперматозоида и яйцеклетки, здоровые родители имеют один шанс из 10,000, родить ребенка с синдромом Марфана. Возможно, в 25% случаев происходит самопроизвольное изменение во время оплодотворения.
Известные люди с синдромом Марфана
Синдром Марфана назван по имени педиатра, который наблюдал девочку с этим заболеванием на протяжении 20 лет. Имеется много интересных фактов о людях, имеющих характерные признаки патологии. Первая манекенщица (Лесли Хорнби – «Твигги»), которая была прототипом для всех чрезмерно худых моделей, болела синдромом Марфана. Наиболее известные личности, о которых есть подобные сведения:

- президент А. Линкольн,
- скрипач Н. Паганини,
- писатель Г. Х. Андерсен,
- композитор С. Рахманинов,
- олимпийский чемпион М. Фелпс.
Распространенность симптомокомплекса у многих одаренных людей дала возможность предположить, что он придает экстраординарные способности.
Патогенез
Синдром Марфана — генетически детерминированное заболевание, характеризующееся поражением или недоразвитием соединительнотканных волокон во время эмбриогенеза и проявляющееся дисфункциональными изменениями со стороны зрительного анализатора, костно-суставной и кардиоваскулярной систем.
Обычно синдром передается по наследству от родителей детям по аутосомно-доминантному принципу. В некоторых случаях он становится результатом мутации гена, кодирующего синтез фибриллина и оказывающего влияние на формирование одновременно нескольких фенотипических признаков.
Генетические изменения – мутации происходят под воздействием негативных эндогенных и экзогенных факторов. Белок фибриллин является важной составной частью многих структур организма. При его недостатке соединительная ткань теряет свою прочность и эластичность, что отражается на состоянии сосудистой стенки и связочно-суставного аппарата.
Синдром Марфана – врожденная аномалия, отличающаяся следующими генетическими признаками:
- аутосомно-доминантным типом наследования, при котором больные встречаются в каждом поколении;
- выраженным плейотропизмом — множественным действием гена;
- варьирующей экспрессивностью – степенью развития признака, контролируемого данным геном;
- высокой пенетрантностью – вероятностью фенотипического проявления признака при наличии соответствующего гена.
Причины
Синдром развивается в результате мутации гена, кодирующего биосинтез особого белка фибриллина — важной структуры межклеточного вещества, которая обеспечивает эластичность и сократительную способность соединительнотканных волокон. Мутация происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде. При недостатке фибриллина нарушается процесс формирования волокон. Он перестают быть прочными и упругими, становятся чрезмерно растяжимыми и менее устойчивыми к деформациям. В наибольшей степени повреждению подвержены сосуды и связки.
Фибриллин необходим для работы цинновой связки, с помощью которой хрусталик прикрепляется к ресничному телу. При дефиците белка эта связка ослабляется, что проявляется миопией, подвывихом хрусталика, вторичной глаукомой, снижением остроты зрения. Кроме зрительного анализатора, белок фибриллин содержится в связках аорты и обеспечивает ее устойчивость к нагрузкам. При ослаблении этих связок происходит расширение сосуда и расслоение его стенок. Подобные изменения являются смертельно опасными для человека. При синдроме Марфана часто отмечается поражение двустворчатого клапана, что требует проведения хирургической коррекции.
Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене FBN1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста — TGFBR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных — первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Симптомы и клинические проявления
Синдром Марфана может проявляться у разных людей по-разному. У некоторых признаки данного типа патологии выражены очень ярко, у других же наблюдаются исключительно лёгкие симптомы. Обычно прогрессирование симптомов происходит по мере того, как человек растёт. Признаки болезни зависят от того, какие системы и органы в организме поражены, поэтому симптоматика может несколько изменяться.

Синдром Марфана характеризуется:
- сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем;
- многообразием проявлений, варьированием сроков появления первых признаков заболевания;
- хроническим прогредиентным течением.
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Вот некоторые основные характеристики людей с синдромом Марфана:
- Скелет — человек с синдромом Марфана обычно очень высокий и худой. В связи с тем, что синдром Марфана сопровождается удлинением костей скелета: туловища, рук, ног, пальцев рук и ног, они могут быть непропорционально длинные. Человек с синдромом Марфана часто имеет длинное, узкое лицо, и его верхняя губа может быть изогнута, причиной чего являются зубы. Другие скелетные аномалии включают изменение грудини (грудной кости), которая или выступает, или зигзагообразной формы, искривление спины (сколиоз) и плоскостопие. Средняя длина тела при рождении у мальчиков с синдромом Марфана составляет 53 см, окончательный рост – 191 см; у девочек — соответственно 52,5 см и 175 см.
- Глаза – у более, чем половины всех людей с синдромом Марфана отмечается смещение одного из двух хрусталиков глаз. Хрусталики глаз могут быть незначительно выше, чем нормальные и смещаться в сторону. Смещение может быть минимальным или резко выраженным и очевидным. Отслоение сетчатки – это серьёзное осложнение. Многие люди с синдромом Марфана близоруки и у них может развиться глаукома (высокое давление внутри глаза) или катаракта.
- Сердце и кровеносные сосуды (сердечно-сосудистая система) – большинство людей с синдромом Марфана имеют аномалии, связанные с сердцем и кровеносными сосудами. Так как имеется дефект соединительной ткани, стенка аорты (большая артерия, которая несет кровь из сердца к телу) может быть ослаблена и растягиваться, этот процесс называют расширением аорты. Расширение аорты — увеличивает риск разрыва аорты, вследствие чего возникает серьёзная проблема с сердцем или иногда внезапная смерть. Иногда могут развиваться дефекты сердечных клапанов. В большинстве случаев, определенные клапаны могут создавать «сердечный шум», который может услышать доктор с помощью стетоскопа. Небольшие нарушения в работе сердца могут не вызывать симптомов, но большие могут стать причиной затруднения дыхания, усталости и сильного сердцебиения (очень быстрого или неправильного сердечного ритма).
- Нервная система – головной и спинной мозг окружены жидкостью; вокруг имеется оболочка, которая называется твердой мозговой оболочкой, ее составляет соединительная ткань. Когда люди с синдромом Марфана стареют, твердая мозговая оболочка часто слабеет и вытягивается. Это называется дуральная эктазия. Эти изменения могут стать причиной легкого дискомфорта или могут привести к боли в брюшной полости или боли, неподвижности или слабости ног.
- Кожа — у большинства людей с синдромом Марфана развивается растягивание кожи, даже без изменения массы тела. Это может появиться в любом возрасте и не представлять опасности. Тем не менее, у людей с синдромом Марфана увеличивается риск развития брюшной или паховой грыжи, при которой развивается выпуклость, которая содержит в себе часть кишки.
- Легкие – хотя аномалии соединительной ткани делают маленькие воздушные мешочки в легких менее эластичным, люди с синдромом Марфана обычно не испытывают существеннных проблем с их легкими. Если, тем не менее, эти маленькие воздушные мешочки удлиняются или распухают, то может увеличиться риск спадения легких. Редко у людей с синдромом Марфана могут возникать нарушения дыхания во время сна, как храп или синдром апноэ (нарушение сна возникает в краткий период, когда дыхание останавливается).
- Самое опасное и скрытое для глаза проявление болезни – это поражение аорты, самой крупной артерии организма, по которой кровь выходит из сердца. У пациентов с синдромом Марфана аорта расширяется и может разорваться. Это может привести к внезапной смерти в молодом возрасте.
Человеку с чертами, напоминающими синдром Марфана, следует пройти обследование у нескольких специалистов: в частности, у окулиста, кардиолога и ортопеда.
Диагностика
Для диагностики синдрома Марфана используются критерии, выработанные в 1986 году американской организацией US Marfan Foundation. Пациент обычно проходит комплексное обследование у хирурга, офтальмолога и кардиолога. Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
Непропорционально развитые конечности, высокий рост, удлиненная голова, длинные “паукообразные” пальцы, деформированная грудная клетка, искривленный позвоночник, ранняя глаукома, отслоение сетчатки, близорукость, астигматизм, дислокация хрусталика, слишком глубоко посаженные глаза — эти признаки могут указывать на заболевание. Также оценивается состояние митрального клапана и корня аорты пациента, наличие или отсутствие страдающих этим синдромом в его роду.
- За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются:
- дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки;
- килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения;
- отношение длины верхнего сегмента тела к нижнему 1,05; сколиоз (> 20˚) или спондилолистез; ограничение разгибания в локтевом суставе (11%).
- Также применяются фенотипические диагностические тесты, определяющие:
- соотношение кисть/рост (при синдроме Марфана > 11%);
- длину среднего пальца (> 10 см);
- индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть 5 см) восходящей части и расслоении аорты.
- ЭКГ при синдроме Марфана позволяет определить:
- нарушение ритма сердца, выраженную гипертрофию миокарда;
- ЭхоКГ — обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты.
- На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца;
- На КТ и МРТ сердца и сосудов — выявить дилатацию и аневризмы аорты.
- Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
- При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Необходимы: УЗИ внутренних органов, биомикроскопия и офтальмоскопия, молекулярно-генетический анализ. Кроме того, общий анализ крови и мочи — визуализируют типичные признаки воспаления, биохимическое исследование крови позволяет выявить дисфункцию определенного органа, возникшую в результате развития патологического процесса, а также определить первопричину заболевания и назначить правильное лечение.
Необходима дифференциальная диагностика с заболеваниями, внешне напоминающими синдром Марфана: гомоцистинурией, врожденной контрактурной арахнодактилией (синдромом Билса), наследственной артроофтальмопатией (синдромом Стиклера), MASS-синдромом, синдромами Элерса-Данлоса, Лойса-Дитца, Шпринцена–Голдберга, семейной эктопией хрусталика и др.
Особенности терапии
Синдром Марфана неизлечим. Этиотропной терапии недуга не существует, ведь невозможно заменить гены ребенка. Больным проводят симптоматическую терапию, целью которой является облегчение общего состояния, устранение симптомов и предупреждение тяжелых осложнений.
Методики лечения болезни:
- приём лекарственных препаратов. В терапию включают антикоагулянты, адреноблокаторы, антиаритмические препараты и прочее;
- хирургическая операция показана в случае наличия пороков сердца, аорты. Возможно проведение протезирования клапанного аппарата;
- нормализация зрения проводится за счёт коррекции миопии, устранения катаракты и глаукомы. Иногда требуется провести имплантацию искусственного хрусталика;
- тракция (при сколиозе);
- операция проводится при поражении позвоночного столба и суставов (пластика суставов, удаление межпозвоночных грыж, протезирование);
- физиолечение и занятие ЛФК;
- если болезнь поразила лёгкие, то проводится их дренирование.
При снижении остроты зрения пациентам с офтальмологическими заболеваниями прописывают очки для постоянного ношения или контактные линзы.
- оперативное вмешательство на сердце и аорте — протезирование аорты и клапанов сердца, реконструктивные и пластические операции,
- операции на глазах — коррекция близорукости лазером, замена хрусталика, устранение глаукомы,
- хирургическая коррекция скелета — пластика грудной клетки при ее воронкообразной деформации, стабилизация позвоночника, протезирование крупных суставов.
Людям с марфаноидным фенотипом стертой формы показано физиолечение и ЛФК.
Прогноз
Прогноз синдрома Марфана неоднозначный. Он зависит от состояния кардиоваскулярной системы, глаз и скелета больного. С помощью диагностических критериев, врачи могут поставить диагноз и назначить лечение, если это требуется.
«В наше время больные синдромом Марфана, если им вовремя поставлен диагноз и назначено лечение, вполне могут дожить до старости. Однако все еще много людей гибнет, потому что их заболевание не было диагностировано», — говорит Кэролин Леверинг, президент Национального Фонда Синдрома Марфана в США.
С помощью своевременной кардиохирургической коррекции можно улучшить качество жизни больных и вернуть им трудоспособность. Если недуг не лечить, продолжительность жизни больных ограничится 30—40 годами. Причиной их смерти становится разрыв аорты или острая коронарная недостаточность. Современная медицина позволяет пациентам успешно лечиться и полноценно жить до самой старости.
Профилактика
Все пары, в семейном анамнезе которых имелись случаи наследственных заболеваний, должны перед беременностью посетить генетика и заранее сдать все необходимы анализы. Пренатальная диагностика — комплекс мероприятий, проводимых с целью выявления патологии на стадии внутриутробного развития. Она заключается в проведении УЗИ плода и биохимического скрининга материнских сывороточных маркеров. К ее инвазивным методам относятся: биопсия ворсин хориона, исследование околоплодных вод, пуповинной крови и клеток плаценты.
Всем больным необходимо вовремя санировать имеющиеся очаги хронической инфекции в организме — лечить кариес, тонзиллит, синусит с помощью антибиотиков. Это крайне важное мероприятие, поскольку у лиц с синдромом Марфана иммунитет ослаблен. Им следует закаляться, правильно питаться, оптимизировать режим дня, полноценно спать, подолгу гулять на свежем воздухе, бороться с вредными привычками, избегать конфликтных ситуаций и стрессов.
Наследование синдрома Марфана в наши дни
Врожденный синдром Марфана передается по аутосомно–доминантному типу. Это значит, что его появление в семье подчиняется таким закономерностям:
- чаще болен один из родителей;
- частота появления у девочек и мальчиков одинаковая;
- вероятность возникновения у ребенка равна 50%;
- от здоровых родителей (при наличии больных бабушек, дедушек) обычно не передается;
- если провести анализ родословного древа, то выявляется вертикальный способ передачи синдрома в каждом поколении;
- не все члены семьи имеют одинаковые признаки болезни.
Следует учитывать, что в 20% случаев мутации гена не связаны с наследственностью, они могут возникать первично. В таких случаях играет роль возраст родителей. Доказано, что чаще дети с этой аномалией развития появляются, если отец старше 35 лет. В следующем поколении больных при таком варианте наследования может и не быть (пропуск поколения), но половина их потомков окажется с этой патологией. Поэтому всем будущим родителям, у которых в семьях были выявлены лица с синдромом Марфана, нужно перед планированием беременности обязательно пройти консультацию в медико-генетическом центре.
Как не пропустить синдром Марфана у детей
Поскольку патология имеет наследственную природу, в большинстве случаев удаётся проследить заболевание в семье. Нужно помнить, что недуг может протекать в различных формах и многие люди, с лёгким течение синдрома, не догадываются о своём заболевании на протяжении всей жизни.
Особенность клинических проявлений
Поскольку болезней, связанных с патологией соединительной ткани, существует множество, иногда бывает сложно правильно поставить диагноз ребёнку. Чтобы справится с этой задачей в 1996 году генетиками и клиницистами были разработаны современные критерии, с помощью которых можно определить синдром Марфана. В основу диагностики были положены «большие» и «малые» признаки заболевания. Их сочетание оценивается специалистом и решается вопрос о наличии генетического синдрома.
Большие критерии
К ним относятся:
- увеличение роста в большей степени за счёт верхней части тела;
- грубая деформация грудной клетки и позвоночника;
- продольное плоскостопие;
- невозможность полностью разогнуть конечность в коленных и локтевых суставах (контрактуры);
- эктопия хрусталика;
- расширение и расслоение восходящей части аорты и другие признаки.
Некоторые из этих симптомов можно встретить у абсолютно здоровых детей. В диагностике генетического синдрома большую роль играет именно их сочетание.
Триада Марфана включает в себя:
- патологию костно-суставной системы,
- органические изменения в сердце или крупных сосудах,
- болезни глаз.
Малые критерии
Эти признаки в меньшей степени указывают на наследственный дефект, но их наличие и сочетание с большими критериями подтверждает диагноз синдром Марфана.
К ним относятся:
- высокая подвижность суставов;
- аномалии зубов, нёба;
- гипоплазия радужной оболочки глаза, цилиарной мышцы, увеличение длины глазного яблока;
- пролапс митрального клапана;
- патологии бронхо-лёгочной системы,
- спонтанный пневмоторакс и другие нарушения.
Диагностические тесты
Для уточнения внешних изменений у ребёнка используются различные способы: измерение роста, длины кисти, соотношение размеров верхней части туловища к нижней и другие. Особо показательны в диагностике синдрома следующие исследования:

- тест запястья. Врач просит ребёнка обхватить запястье одной руки большим пальцем и мизинцем другой руки, образуя «браслет». В пользу наследственного недуга говорит лёгкое смыкание кисти на запястье другой руки, нахождение фаланг мизинца и большого пальца друг на друга;
- тест большого пальца. Исследователь просит малыша попытаться дотянутся большим пальцем до предплечья этой же руки. Тест считается положительным, если ногтевая фаланга пальца ребёнок с лёгкостью достаёт до лучевой кости предплечья.
Лабораторные исследования
Обычные клинические и биохимические анализы крови и мочи не показательны при синдроме Марфана, изменений в них может не быть. Помочь в установлении диагноза поможет обнаружение продуктов метаболизма соединительной ткани в моче. Резкое увеличение оксипролина и гликозаминогликанов в суточной моче может говорить о развитии осложнений у ребёнка (прогрессировании сердечной недостаточности, отслойке плаценты, развитии пневмоторакса, пневмонии).
С помощью современных методов исследования, молекулярно-генетической диагностики можно обнаружить характерную для данного генетического синдрома мутацию в гене FBN1.
Такое заболевание, как синдром Билса отличается от синдрома Марфана дефектом синтеза другого белка – фибриллина 2, но клинические симптомы этих недугов схожи. Различить их можно только с помощью молеклярно-генетической диагностики и выявления особого симптома «мятого уха», что указывает на наличие синдрома Билса.
Источник https://www.krasotaimedicina.ru/diseases/children/marfan-syndrome
Источник https://medportal.ru/enc/rheumatology/systemic/sindrom-marfana/
Источник https://clinic-a-plus.ru/articles/sosudi-veni/2143-chto-takoe-sindrom-marfana-opisanie-patologii.html