Фенилкетонурия
Обновлено: 08.06.2023 Фенилкетонурия (ФКУ) – достаточно тяжелое наследственное заболевание, при котором нарушение обмена аминокислот приводит к их накоплению в клетках организма. Характеризуется поражением головного мозга. При отсутствии лечения ФКУ может привести к умственной отсталости, судорогам, поведенческим проблемам и психическим расстройствам. Ребенок, рожденный от матери, с признаками ФКУ, может иметь проблемы с сердцем, маленькую голову и низкий вес при рождении.
Симптомы ФКУ
- светлая кожа и светлые волосы (обусловлены сниженной выработкой пигмента меланина);
- сыпь, напоминающая экзему, включающая аллергическое воспаление после выделения кетонов (атопический дерматит);
- чувствительность к солнечному свету (фоточувствительность) – красная или розовая кожная сыпь после воздействия солнечного света;
- увеличение частоты гнойных инфекций;
- образование ороговевших участков в волосах, на тыльной стороне рук, ягодицах и передней части бедер (кератоз пиларис);
- уменьшение доли пигментных родинок (пигментных невусов);
- склероз соединительной ткани кожи (склеродермические бляшки) – заболевание, вызывающее отвердение и сужение кожи.
Следствием заболевания является тяжелая умственная отсталость и микроцефалия. При отсутствии терапевтических мер развиваются такие поведенческие расстройства, как самоповреждение, крайняя гиперактивность и психические припадки. Не исключен паркинсонизм (экстрапирамидные проявления) и субпигментация в радужной оболочке глаз.
Причины возникновения фенилкетонурии
Ген PAH, мутация которого вызывает развитие заболевания, кодируется с образованием белка, называемого фенилаланиновой гидроксилазой. Этот белок на самом деле является ферментом, задачей которого является преобразование аминокислоты фенилаланина в тирозин, другую аминокислоту.
Поскольку фермент не вырабатывается нормально, на медицинском языке формируется состояние, называемое гиперфенилаланинемией или ГПА. Параллельно с повышением уровня аминокислоты фенилаланина, что приводит к серьезному повреждению центральной нервной системы, образуется острый дефицит тирозина, продукта превращения фенилаланина.
Тирозин превращается в биохимическом процессе в меланин, пигмент, который придает темный цвет глазам, волосам и коже. Так, среди страдающих этим заболеванием, обычно можно увидеть очень светлые глаза, светлые волосы и кожу.
Дефектный ген передается аутосомно-рецессивно, поэтому для того, чтобы болезнь появилась у потомства, оба родителя должны быть носителями дефектного гена и оба должны передать дефектный ген потомству.
Методы диагностики ФКУ
Сегодня тест на фенилкетонурию делают в роддоме всем новорожденным через 2 дня после появления на свет. Это обследование должно своевременно выявить больных детей, чтобы назначить соответствующее лечение. Диагностика ФКУ включает:
Фенилкетонурия
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Фенилкетонурия: причины появления, симптомы, диагностика и способы лечения.
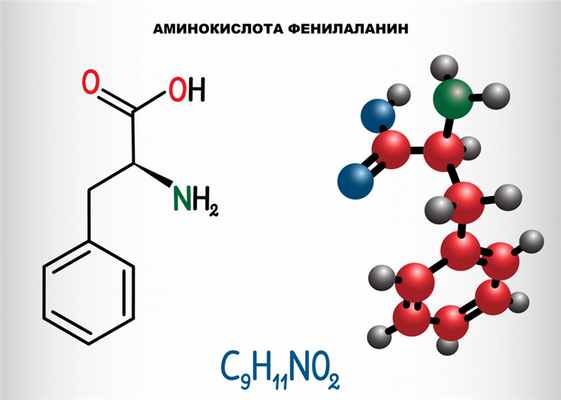
Фенилкетонурия (ФКУ) – группа аутосомно-рецессивных заболеваний, обусловленных нарушением обмена незаменимой аминокислоты фенилаланина, поступающей в организм человека с белковой пищей.
Причины появления фенилкетонурии
Причина заболевания связана с нарушением обмена незаменимой аминокислоты фенилаланина, приводящим к повышению ее уровня в крови, тканях и биологических жидкостях.
Избыток фенилаланина токсичен для нервной системы и при длительном воздействии вызывает в ней необратимые дегенеративные изменения.
Фенилаланин не синтезируется в клетках организма и поступает только с пищей. Метаболизм этой аминокислоты осуществляется двумя путями: участвует в биосинтезе белка или превращается в тирозин под действием фермента фенилаланингидроксилазы. В клетках меланоцитах, которые присутствуют в коже, волосах, сетчатке глаза, тирозин превращается в пигмент меланин. В щитовидной железе из тирозина синтезируются гормоны тироксин и трийодтиронин. В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов – дофамина, норадреналина, адреналина.
Фенилкетонурия объединяет несколько форм нарушения обмена фенилаланина, сходных по клиническим признакам:
- классическая (ФКУ I типа), обусловленная мутацией гена ФАГ (РАН) фенилаланингидроксилазы;
- ФКУ II типа, обусловленная мутацией структурного гена для цитозольной дигидроптеридинредуктазы, что вызывает метаболический блок на путях превращения фенилаланина в тирозин, а также предшественников нейромедиаторов катехоламинового и серотонинового ряда;
- ФКУ III типа вызвана мутацией структурного гена для цитозольной пирувоилтетрагидроптерин синтазы, что приводит к ее недостаточности в печени и эритроцитах.
По уровню фенилаланина в сыворотке крови выделяют:
- легкую гиперфенилаланинемию – 120-600 мкмоль/л (2−10 мг/дл);
- умеренную (мягкую, среднюю) ФКУ – 600-1200 мкмоль/л (10−20 мг/дл);
- классическую (тяжелую) ФКУ – 1200 мкмоль/л и более (>20 мг/дл).
Новорожденный с ФКУ выглядит здоровым, а первые симптомы манифестируют при употреблении в пищу белковой пищи, содержащей большое количество фенилаланина.
Проявления заболевания зависят от тяжести мутационного повреждения фермента, степени активности фермента и сроков начала терапии.
Классическая фенилкетонурия характеризуется полным или почти полным дефицитом фенилаланингидроксилазы в печени. Проявляется у ребенка в возрасте 2-6 месяцев вялостью, отсутствием интереса к окружающему, иногда повышенным беспокойством, срыгиванием, снижением мышечного тонуса, признаками атопического дерматита, задержкой психомоторного развития, судорогами.
Эпилептические приступы могут быть первым признаком болезни и встречаются почти у половины нелеченных больных. Приступы носят упорный характер и плохо поддаются терапии.
Во втором полугодии жизни дети перестают реагировать на обращенную к ним речь, узнавать родителей, не фиксируют взгляд и не реагируют на яркие игрушки, не переворачиваются на живот, не сидят. Отмечается задержка статико-моторного и психоречевого развития.
Нарушения обмена аминокислот приводят к гипопигментации волос и кожи, поскольку у детей почти не вырабатывается пигмент меланин. В результате присутствует гиперчувствительность к инсоляции, может наблюдаться тяжелая экзема, дерматит, фолликулярный кератоз, повышенная склонность к гнойничковым инфекциям. У больных отмечают «мышиный» запах мочи, который объясняется выделением метаболитов фенилаланина. При отсутствии лечения медленно прогрессирует умственная отсталость. Могут наблюдаться двигательные, психопатоподобные и шизофреноподобные расстройства.
Умеренная фенилкетонурия характеризуется низкой остаточной ферментативной активностью. Клинически болезнь проявляется на первом-втором году жизни и медленно прогрессирует при отсутствии лечения.
Клинические проявления ФКУ II сходны с классической фенилкетонурией. Однако есть и свои особенности: гибель нейронов, кальцификация и анормальная васкуляризация центральной коры, белого вещества, базальных ганглий и таламуса, а также нарушение метаболизма фолатов. В клинической картине преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, мышечная дистония (непроизвольные движения и формирование патологических поз), спастический тетрапарез (комплекс двигательных нарушений верхних и нижних конечностей).
Проявления фенилкетонурии III напоминают болезнь Паркинсона. Наблюдаются затруднения в удержании равновесия в определенной позе или при смене позы, недостаточная двигательная активность с ограничением скорости и объема движений, увеличение секреции слюнных желез, нарушения глотания, а также тяжелая умственная отсталость, микроцефалия.
Игнорирование рекомендаций по диетотерапии, недостаточный контроль уровня фенилаланина в крови могут иметь отдаленные последствия: низкий коэффициент интеллекта, замедленная речь, нарушения памяти, проблемы с концентрацией внимания и поведением.
У взрослых пациентов, прекративших соблюдение диеты, наблюдается ухудшение неврологического и психологического состояния, возникает поздняя эпилепсия, атаксия, тремор, депрессивные расстройства, неврозы.
Если пациенты соблюдают диету с ограничением высокобелковых продуктов, но не принимают специальные аминокислотные смеси без фенилаланина, существует риск развития дефицита витаминов, микро- и макроэлементов.
Женщинам с ФКУ до зачатия и во время беременности необходимо строго придерживаться рекомендованного врачом диетического питания, в противном случае у родившихся детей может диагностироваться синдром материнской фенилкетонурии, который проявляется дисморфией лица, задержкой умственного и физического развития, микроцефалией (значительным уменьшением размера черепа), врожденными пороками сердца.
Определить риск рождения ребенка с ФКУ можно с помощью генетической экспертизы у обоих родителей до зачатия ребенка:
- фенилкетонурия, PAH м.;
Исследование мутаций в гене PAH. Фенилкетонурия (гиперфенилаланинемия, ФКУ, ГФА, OMIM261600) — группа гетерогенных аутосомно-рецессивных заболеваний, обусловл�.
Фенилкетонурия (ФКУ)
Фенилкетонурия это нарушение метаболизма аминокислот Обзор нарушений метаболизма аминокислот и органических кислот (Overview of Amino Acid and Organic Acid Metabolism Disorders) Почки активно реабсорбируют значительные количества аминокислот. Дефекты переноса аминокислот в почечной канальце включают цистинурию и болезнь Хартнупа, которые обсуждаются в другом месте. Прочитайте дополнительные сведения , приводящее к возникновению клинического синдрома умственной отсталости с когнитивными и поведенческими расстройствами, вызванными повышенным уровнем фенилаланина. Основной причиной является недостаточность активности фенилаланин гидроксилазы. Диагноз ставят на основании выявления высокого уровня фенилаланина при нормальном или низком содержании тирозина. Лечение – пожизненная диета без фенилаланина. Прогноз при лечении отличный.
Для получения информации о других нарушениях аминокислот, см. таблицу нарушения обмена фенилаланина и тирозина Нарушения метаболизма фенилаланина и тирозина . См. также Подходы к лечению пациентов с подозрением на наследственное нарушение обмена веществ (Approach to the Patient With a Suspected Inherited Disorder of Metabolism) Подходы к лечению пациентов с подозрением на наличие наследственного заболевания обмена веществ Большинство наследственных нарушений метаболизма (врожденные нарушения обмена веществ) являются редкими, и, следовательно, для их диагностики необходим более высокий индекс подозрения. Своевременная. Прочитайте дополнительные сведения .
Патофизиология фенилкетонурии
Избыточно потребляемый фенилаланин (т.е. не использующийся для синтеза белка), как правило, преобразуется в тирозин фенилаланингидроксилазу; тетрагидробиоптерин (BH4) является необходимым кофактором в данной реакции. Когда одна из нескольких мутаций гена приводит к недостаточности или отсутствию фенилаланингидроксилазы, происходит накопление потребляемого фенилаланина; мозг является основным повреждаемым органом, возможно, из-за нарушения миелинизации.
Некоторый избыток фенилаланина метаболизируется в фенилкетоны, которые выводятся из организма с мочой, что приводит к развитию фенилкетонурии. Степень дефицита фермента и, следовательно, тяжести гиперфенилаланинемии варьируется среди пациентов в зависимости от конкретной мутации.
Варианты заболевания
Хотя почти все случаи (98–99%) ФКУ развиваются в результате дефицита фенилаланингидроксилазы, фенилаланин также может накапливаться, если BH4 не синтезируется из-за недостаточности дигидроптерин синтазы или не регенерируется из-за недостаточности дигидропиридин редуктазы. Кроме того, поскольку BH4 также является кофактором тирозингидроксилазы, участвующей в синтезе дофамина и серотонина, дефицит BH4 изменяет синтез медиаторов, вызывающих неврологические симптомы независимо от накопления фенилаланина.
Симптомы и признаки фенилкетонурии
Большинство детей с фенилкетонурией при рождении нормальные, симптомы и признаки появляются постепенно, в течение нескольких месяцев, по мере накопления фенилаланина. Отличительная черта нелеченой ФКУ – тяжелая умственная отсталость. У детей также возникают крайняя гиперактивность, нарушение походки, психозы, часто возникает неприятный «мышиный» запах тела, обусловленный фенилуксусной кислотой (продукт распада фенилаланина) в моче и поте. Кроме того, дети, как правило, имеют более светлую кожу, волосы и цвет глаза, чем непоражённые члены семьи, и у некоторых из них болезнь может проявляться сыпью, похожей на младенческую экзему.
Диагностика ФКУ
Рутинный неонатальный скрининг
(См. также the American College of Medical Genetics and Genomics Therapeutic Committee’s 2013 diagnosis and management guidelines for phenylalanine hydroxylase deficiency.)
В США и многих развитых странах, всем новорожденным проводят скрининг на фенилкетонурию Скрининговые тесты для новорождённых Рекомендации по скринингу новорожденных зависят от клинических условий и государственных стандартов. Определение группы крови показано, если мать имеет кровь группы 0 или резус-отрицательную. Прочитайте дополнительные сведения в течение 24-48 часов после рождения, проводя один из нескольких анализов крови Начальное тестирование Большинство наследственных нарушений метаболизма (врожденные нарушения обмена веществ) являются редкими, и, следовательно, для их диагностики необходим более высокий индекс подозрения. Своевременная. Прочитайте дополнительные сведения ; отклоняющиеся от нормы результаты подтверждают непосредственным измерением уровня фенилаланина. В классической ФКУ уровни фенилаланина у новорожденных часто составляют > 20 мг/дл (1,2 ммоль/л). Те, у кого есть частичный недостаток, как правило, имеют уровни 8–10 мг/дл, в то время как на обычной диете уровни > 6 мг/дл уже требуют лечения; для отличия от классической ФКУ требуется анализ мутаций, идентифицирующий незначительные мутации гена или, реже, измерение активности фенилаланин-гидроксилазы печени, которое показывает уровень активности в 5–15% от нормального.
Дефицит BH4 отличается от других форм ФКУ повышенными концентрациями биоптерина или неоптерина в моче, крови, спинномозговой жидкости или во всех 3 жидкостях; также можно использовать генетическое тестирование. Распознавание важно, биоптериновый профиль мочи при первичной диагностике нужно определять регулярно, поскольку стандартное лечение ФКУ не предотвращает неврологические повреждения.
Диагностику у детей в семьях с положительным семейным анамнезом можно проводить пренатально с помощью прямого исследования мутации после биопсии хориона или амниоцентеза.
Прогноз при ФКУ
Адекватное лечение, начатое в первые дни жизни, предотвращает возникновение тяжелых проявлений заболевания. Тем не менее умеренный когнитивный дефицит и проблемы с психическим здоровьем все еще могут возникать даже при хорошем диетическом контроле. Лечение, начатое после 2–3 лет, может быть эффективно только в контроле крайней гиперактивности и некупируемых судорог.
Дети, рожденные от матерей с плохо контролируемой ФКУ (т.е. имеющих высокий уровень фенилаланина), во время беременности имеют высокий риск микроцефалии и дефектов развития.
Лечение ФКУ
Ограничение потребления фенилаланина
Лечение фенилкетонурии заключается в соблюдении пожизненной диеты без фенилаланина. Все природные белки содержат около 4% фенилаланина. Поэтому основные продукты питания включают
Натуральные продукты с низким содержанием белка (например, фрукты, овощи, некоторые злаки)
Белковые гидролизаты, из которых удален фенилаланин
Смеси аминокислот без фенилаланина
Примерами коммерчески доступных продуктов без фенилаланина являются продукты PKU Anamix ® (для младенцев), XPhe Maxamaid ® (для детей от 1 до 8 лет), XP Maxamum ® (для детей > 8 лет); Phenex ® -1 и Phenex ® -2; Phenyl-Free ® 1 и Phenyl-Free ® 2; PhenylAde ® (различные); PKU Lophlex ® LQ; и Phlexy-10 ® (различные формы). Некоторое количество фенилаланина необходимо для роста и метаболизма; эта потребность удовлетворяется за счет измеряемых количеств природного белка с молоком или фруктами с низким содержанием белка.
Требуется частый мониторинг уровней фенилаланина в плазме; рекомендуемые значения для всех детей располагаются в пределах 2–6 мг/дл (120–360 мкмоль/л). Планирование диеты и управление ею должно войти в привычку у женщин детородного возраста до беременности – так они помогут своим будущим детям. Тирозин используют чаще, поскольку это незаменимая аминокислота у пациентов с ФКУ. Кроме того, всем пациентам с дефицитом фенилаланин-гидроксилазы необходимо провести пробное назначение сапроптерина, назначение которого может иметь преимущества.
Для лиц с BH4-недостаточностью лечение также включает тетрагидробиоптерин по 1–5 мг/кг перорально 3 раза в день, леводопу, карбидопу и 5-OH триптофан и фолиевую кислоту по 10–20 мг перорально 1 раз в день при дефиците дигидроптерин-редуктазы. Тем не менее цели лечения и подход являются такими же, как при ФКУ.
Ключевые моменты
ФКУ возникает как последствие, когда одна из нескольких мутаций гена приводит к недостаточности или отсутствию фенилаланингидроксилазы, вслседствие чего происходит накопление потребляемого с пищей фенилаланина; мозг является основным повреждаемым органом, возможно, из-за нарушения миелинизации.
Фенилкетонурия (ФКУ) вызывает клинический синдром умственной отсталости с когнитивными и поведенческими нарушениями; при отсутствии лечения развивается тяжелая умственная отсталость.
В США и многих развитых странах, всех новорожденных обследуют на фенилкетонурию в течение 24-48 часов после рождения, проводя один из нескольких анализов крови; отклоняющиеся от нормы результаты подтверждают непосредственным измерением уровня фенилаланина.
Лечение заключается в пожизненном ограничении поступления фенилаланина с пищей; адекватное лечение, начатое в первые дни жизни, предотвращает возникновение многих проявлений этого заболевания.
Несмотря на то, что при наличии лечения прогноз благоприятный, требуется частый мониторинг уровней фенилаланина в плазме; рекомендуемые значения располагаются в пределах 2–6 мг/дл (120–360 мкмоль/л) для всех детей.
Дополнительная информация
Ниже следуют англоязычные ресурсы, которые могут быть информативными. Обратите внимание, что The manual не несет ответственности за содержание этих ресурсов.
American College of Medical Genetics and Genomics Therapeutic Committee: Diagnosis and management guidelines for phenylalanine hydroxylase deficiency (2013)
Online Mendelian Inheritance in Man® (OMIM®) database: полная информация о генах и их молекулярной и хромосомной локализации
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Фенилкетонурия
Фенилкетонурия – наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных. В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты.
Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным.
Скрининг-тест проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Прогноз и профилактика фенилкетонурии
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Фенилкетонурия

Фенилкетонурия (либо ФКУ) — это наследственное, редко встречающееся заболевание, которое проявляется в нарушении обмена аминокислот. Если точнее, то организм людей с такой болезнью не может расщеплять фенилаланин, поступающий вместе с белковой пищей. Из-за такого дисбаланса в теле накапливаются опасные соединения, способные отравлять нервную систему, в том числе и ткани головного мозга. Эти отравления могут быть настолько сильными, что у ребенка развивается умственная отсталость, включая идиотию.
Несмотря на серьезную опасность болезни, сейчас ее вполне можно нейтрализовать — то есть дети, которые рождаются с такой проблемой, не обречены.
Статистика
Количество детей с таким заболеванием зависит от страны, местности проживания. Так, в России фиксируют рождение одного малыша, страдающего фенилкетонурией, на 10 000 человек. В Великобритании этот показатель в два раза выше, а у африканских детей подобная проблема почти не встречается. Девочки страдают этим недугом чаще — почти в два раза по сравнению с мальчиками.
Симптомы
У детей с ФКУ при рождении нет характерных симптомов, которые позволят сразу понять, что с малышом что-то не так. Ребенок с такой проблемой выглядит абсолютно здоровым. И если такой малыш употребляет в пищу белковую еду с большим количеством фенилаланина, начинаются первые симптомы. По этой причине диагностика проводится массово, всем детям сразу после рождения (см. ниже).
Если по какой-то причине диагностика не была проведена, и ребенок с болезнью получает белок, со временем у него начнутся тревожные симптомы. Первыми будут слабость, беспокойство, отсутствие у малыша улыбки, ярких реакций на окружающий мир. Дальше идут симптомы медленного развития: например, когда приходит время, ребенок не пытается сесть, перестает узнавать маму и т. д.

По мере развития болезни могут проявляться такие симптомы фенилкетонурии:
- Изменение цвета волос и глаз к более светлым. Это связано с тем, что в организме недостаточно меланина.
- Прибавление веса — малыш начинает быстро поправляться.
- Высыпания, экзема, сухость и шелушение кожи.
- Частая рвота.
- Неприятный («мышиный») запах мочи
К двум-трем годам основные диагностические признаки фенилкетонурии усиливаются и выглядят уже так:
- Спазмы и судороги, тремор пальцев рук.
- Постоянно зажатая поза. Ребенок не может расслабиться из-за сильного напряжения в мышцах.
- Неадекватные действия. Так, малыш может резко засмеяться, закричать — то есть просто ведет себя неадекватно ситуации.
- Деформация ушей, уменьшение черепа в отношении размеров тела.
- Выступающая нижняя челюсть.
- Недержание.
Причины
Поскольку речь идет о генетическом заболевании, причины фенилкетонурии сводятся исключительно к мутации гена 12-й хромосомы. Именно этот ген отвечает за определенный фермент, помогающий перерабатывать аминокислоту фенилаланин. Из-за мутации количество фермента становится меньше, поэтому сама аминокислота накапливается в тканях, отравляя их. Все это влияет и на нейромедиаторы, работу нервных проводников.
Заболевание возникает тогда, когда патологические гены матери и отца совпадают, оно не зависит от пола ребенка.
Названная причина фенилкетонурии определяет развитие болезни в 98% случаев, однако есть еще 2%, когда проблема заключается в других генетических дефектах. Если в первом случае (почти всегда) назначается лечение диетой — и оно очень эффективно, то во второй ситуации оно будет бесполезным.
Диагностика
Определить риски появления этого заболевания можно еще до зачатия ребенка — если оба родителя пройдут генетическую экспертизу. Но после рождения малыша его обязательно обследуют на наличие фенилкетонурии. В России это обязательная процедура, которая входит в неонатальный скрининг. Анализ обязательно нужно взять в течение первых трех недель жизни, так что, если малыш появился на свет не в роддоме, родителям обязательно нужно об этом позаботиться. Результаты анализа можно получить через сутки.
Если в анализе обнаружен ген с изменениями, то малыша и родителей обследуют в медико-генетическом центре. Есть ситуации, когда диагноз после тщательного изучения всех данных опровергается. Но чтобы подтвердить его или убрать, необходимо провести дополнительную диагностику фенилкетонурии:
- изучить сыворотку и сухое пятно крови,
- провести потовый тест,
- сделать копрограмму,
- провести ДНК-диагностику.
Затягивать с рекомендациями врачей нельзя ни в коем случае. Занимаются лечением педиатры и эндокринологи.
Лечение
Основное лечение фенилкетонурии сводится к тому, чтобы убрать из рациона ребенка животные белки. Если это будет сделано в первые недели жизни, малыш будет развиваться нормально — и умственная отсталость ему не грозит. Чем позже будет пересмотрен рацион, тем выше риски, что процесс будет запущен, а потому его можно будет только затормозить.
Строгая диета соблюдается больными в среднем до 18 лет — после этого возраста белки допускаются, но их количество необходимо контролировать. Если в последующем девочка с таким диагнозом вырастет и захочет родить, до зачатия, а также во время беременности и лактации ей нужно будет вернуться к строгой диете без белка. Чтобы восполнить питательные вещества, которые ребенок не может получить из пищи, ему нужно заменить белковую еду специальными продуктами с пептидами и свободными аминокислотами. Для этого есть целенаправленно разработанные решения, с которыми родителей ознакомят врачи. Также при лечении фенилкетонурии можно кормить малышей грудным молоком, но маме придется тоже следить за питанием и сидеть на специальной диете.
Диета ребенка зависит и от возраста. Если сначала важно исключить животный белок, то в дошкольном и школьном возрасте из рациона убирается любой белок. Строго необходимо соблюдать и возрастные нормы фенилаланина.
Поскольку основные диагностические признаки фенилкетонурии нельзя определить визуально, а только по анализам крови, детям с таким диагнозом нужно регулярно сдавать кровь и контролировать количество фенилаланина. Нормой в данном случае являются показатели 180-240 мкмоль/л либо 3-4 мг%. Регулярность анализов также зависит от возраста. До трех месяцев ребенка проверяют каждую неделю, затем постепенно количество визитов к врачу снижается.
Если у вашего ребенка выявлена классическая фенилкетонурия, и вы хотите получить консультацию опытного эндокринолога, а также сдать очередные анализы, клиника АО «Медицина» в Москве — место, где вам обязательно помогут. Большой опыт лечения детей с самыми разными недугами позволяет профессионалам подбирать оптимальные программы восстановления и поддержания для каждого малыша.

Профилактика
Поскольку речь идет о генетическом заболевании, специфической профилактики фенилкетонурии просто не существует. Родители могут сдать специальный анализ до зачатия и посмотреть, каковы риски, что у них родится ребенок с таким недугом. Но независимо от того, был ли пройден генетический тест, всех новорожденных малышей в роддоме все равно проверяют.
Единственная профилактическая мера касается тех родителей, которые по каким-то причинам приняли решение рожать в домашних условиях или в странах, где проверка на фенилкетонурию не проводится обязательно. Им необходимо в течение первых трех недель жизни малыша сдать анализы крови и проверить новорожденного на наличие искаженного 12-го гена.
Вопросы и ответы
Как влияет фенилкетонурия на умственное развитие ребенка?
Если вовремя сесть на диету (с первых недель жизни малыша), то заболевание никак не повлияет на развитие. При грамотном лечении ребенок вырастет полноценным и сможет учиться, социализироваться — то есть не почувствует на себе никаких ограничений кроме тех, что связаны с диетой.
Что будет, если начать диету значительно позже?
К сожалению, задержка в развитии в этом случае не компенсируется. В ряде случаев ее можно задержать — и ребенок будет жить достаточно комфортно. Но очень часто это лишь замедление патологических процессов, так что своевременное лечение просто необходимо.
Можно ли избежать фенилкетонурию?
Поскольку заболевание обусловлено генетически, избежать его, когда встречаются два «поломанных» гена, невозможно. Родители могут только предотвратить проблемы, вызванные болезнью. Но сделать так, чтобы малыш точно не заболел, на данном этапе развития медицины невозможно.
Читайте также:
- Смешанная и атактическая алкогольная полиневропатия. Акупунктура при полиневропатии
- Покраснение глаз
- Аспергиллез. Возбудитель аспиргиллеза. Клиника аспиргиллеза.
- Первая медицинская помощь
- Даптомицин
Фенилкетонурия — симптомы и лечение
Что такое фенилкетонурия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Алексенцевой Елены Сергеевны, педиатра со стажем в 14 лет.
Над статьей доктора Алексенцевой Елены Сергеевны работали литературный редактор Юлия Липовская , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина

Врач УЗИ Cтаж — 14 лет
Перинатальный центр
Дата публикации 16 марта 2020 Обновлено 29 ноября 2021
Определение болезни. Причины заболевания
Фенилкетонурия (ФКУ) — генетическое заболевание, в основе которого лежит врождённое нарушение метаболизма аминокислот, характеризующееся повышенным содержанием фенилаланина в крови. Это аутосомно-рецессивная патология, т. е. ребёнок может унаследовать данное заболевание только в том случае, если оба родителя являются носителями дефектной версии гена.
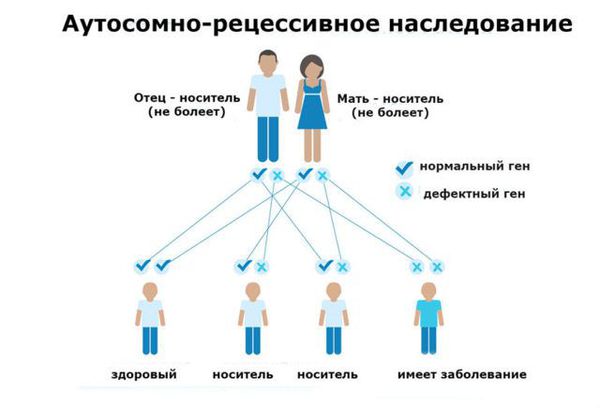
ФКУ связана с дефектом в гене PAH (Phenylalanine hydroxylase gene). Этот ген участвует в образовании фенилаланин-гидроксилазы — печёночного фермента, ответственного за расщепление фенилаланина. Из-за дефекта гена количество этого фермента снижается, а уровень фенилаланина в организме увеличивается, что влечёт за собой такие тяжёлые и необратимые последствия, как умственная отсталость глубокой степени и эпилептические приступы, плохо отвечающие на стандартную антиконвульсантную терапию. П ытаясь избавиться от избытка фенилаланина и его метаболитов ( фенилпировиноградной и фенилуксусной кислот), организм выводит их с мочой .
Распространённость фенилкетонурии
Распространённость фенилкетонурии широко варьируется во всём мире. В Европе частота встречаемости данного заболевания составляет 1 случай на 10 000 живорождённых детей, но для некоторых районов она значительно выше, что связано с большим количеством близкородственных браков. Например, в Турции фенилкетонурия выявляется примерно у одного из 4000 живорождённых детей [1] . В Финляндии, по данным статистики, зарегистрирована самая низкая частота ФКУ среди Европейских стран: 1 случай на 100 000 детей [2] . Что касается нашей страны, усреднённый показатель распространённости данного заболевания — 1 случай на 7000 живорождённых детей.
Факторы риска фенилкетонурии
Основной фактор риска фенилкетонурии — это наличие у обоих родителей дефекта в гене PAH (Phenylalanine hydroxylase gene). Заболевание развивается, если оба родителя передают ребёнку копию повреждённого гена.
Другим ф актором развития фенилкетонурии можно назвать этническую принадлежность ребёнка. Считается, что у афроамериканцев данное заболевание встречается реже, чем у других этнических групп [22] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы фенилкетонурии
Как проявляется фенилкетонурия у новорождённых
У новорождённых с фенилкетонурией симптомы изначально отсутствуют и развиваются медленно, в течение нескольких месяцев, что связанно с постепенным накоплением в организме фенилаланина [3] . Выявить заболевание можно только с помощью неонатального скрининга.
Чем дольше ребёнок с фенилкетонурией не получает специфического лечения, тем быстрее развивается умственная отсталость и необратимые нарушения развития. Кроме того, для детей с фенилкетонурией характерны следующие признаки [4] :
- специфический неприятный запах изо рта, от кожи или мочи, который можно сравнить с затхлым «мышиным» запахом;
- приступы тошноты и рвоты;
- светочувствительность;
- неврологические нарушения, которые нередко могут включать эпилепсию (50 %);
- экстрапирамидные нарушения (паркинсонизм — неврологический синдром, который характеризуется тремором в состоянии покоя, мышечной ригидностью и общей замедленностью движений);
- нарушения зрения: косоглазие, атрофия зрительного нерва, снижение остроты зрения;
- кожные высыпания по типу экземы, локализующиеся на любых участках тела;
- светлая кожа и голубые глаза (из-за невозможности фенилаланина превращаться в меланин);
- микроцефалия (уменьшение размеров черепа и головного мозга при нормальных размерах других частей тела);
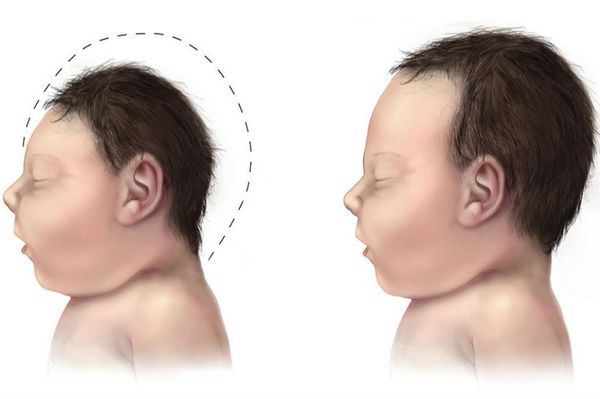
- гиперактивность: беспокойство, неконтролируемые вспышки гнева, невозможность сосредоточиться на выполняемых задачах, спонтанное снижение концентрации;
- интеллектуальная недееспособность: от лёгкой недостаточности интеллекта до идиотии и имбецильности;
- проблемы в поведенческой, эмоциональной и социальной сферах: трудности обучения в образовательных учреждениях, снижение мотивации при выполнении рабочих задач, снижение социальных коммуникаций, раздражительность, перепады настроения, социальная изоляция;
- психические расстройства: депрессии, генерализованные тревоги, шизофрения, биполярное расстройство.
У детей, рождённых от матерей с фенилкетонурией и неконтролируемым уровнем фенилаланина в крови, риск умственной отсталости значительно выше , поскольку они подвергаются воздействию очень высоких и токсичных уровней фенилаланина ещё до рождения. Такие дети могут иметь низкий вес при рождении, что вызывает проблемы прибавки массы и физического развития в будущем. Кроме того, умственно они развиваются значительно медленнее, чем их здоровые сверстники. Другими характерными признаками являются различные пороки сердечно-сосудистой системы (тетрада Фалло, коарктация аорты, дефект межжелудочковой перегородки, дефект межпредсердной перегородки) и поведенческие проблемы, связанные в будущем с нарушением социальной адаптации [5] .
Патогенез фенилкетонурии
Фенилкетонурия как самостоятельное заболевание было открыто норвежским врачом Иваром Асбьёрном Фёллингом ещё в 1934 году. Несмотря на это, вопрос о патогенезе долгое время оставался открытым.
Фенилаланин — это незаменимая аминокислота, которая участвует с синтезе белков. Незаменимая она потому, что организм не может самостоятельно её синтезировать, фенилаланин можно получить исключительно из пищи (мясных и рыбных продуктов, творога, сыра, яйц, орехов, хлебобулочных изделий, круп) или с помощью протеолиза — процесса гидролиза белков с помощью ферментов-протеаз.
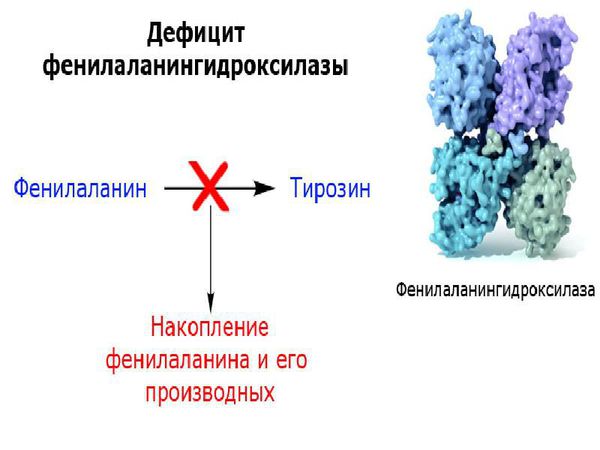
Основной метаболический путь фенилаланина включает его преобразование до тирозина под действием фермента фенилаланин-гидроксилазы, который обнаруживается в печени и почках. Тирозин — это аминокислота, которая участвует в образовании таких важных соединений, как нейромедиаторы (дофамин, адреналин и норадреналин) и пигмент меланин. Нейромедиаторы передают электрические импульсы между нервными клетками (нейронами) и от нейронов к другим клеткам (например мышечным или железистым), что имеет немаловажную роль в когнитивной деятельности. Пигмент меланин защищает организм человека от воздействия ультрафиолетовых лучей [6] .
У пациентов, страдающих фенилкетонурией, из-за дефекта гена и недостатка фермента фенилаланин-гидроксилазы происходит увеличение в плазме крови концентрации фенилаланина (более 1200 мкмоль/л при норме 0-120 мкмоль/л ) и его метаболитов. Одновременно с этим снижается уровень тирозина и его производных (дофамина, адреналина, норадреналина и меланина). Такое состояние оказывает выраженное нейротоксическое действие на структуры мозга. Если пациент с фенилкетонурией не получает или не соблюдает лечение, то у него отмечаются повреждения мозолистого тела, полосатого тела, изменения коры и гипомиелинизация (снижение содержания миелина, образующего оболочку нервных волокон, в различных структурах оболочек мозга). Эти изменения могут привести к снижению интеллектуального развития и нейродегенерации — прогрессирующей гибели нервных клеток. Поэтому пациенты с фенилкетонурией более восприимчивы к нарушениям, связанным с дефицитом дофамина в головном мозге, таким как паркинсонизм.
Хотя патофизиологические механизмы повреждения головного мозга у пациентов с фенилкетонурией ещё не совсем понятны, существует множество свидетельств метаболических изменений, которые включают:
- дефицит биоэнергетики (нарушение процессов преобразования энергии в организме человека);
- окислительный стресс;
- нарушение метаболизма липидов и белков;
- нарушение оптимального уровня кальция и синтеза нейромедиаторов в головном мозге.
Окислительный стресс — это повреждение клеток активными формами кислорода, которые представляют собой молекулы с повышенной реактивностью из-за наличия неспаренного электрона на внешнем электронном уровне. Активные формы кислорода образуются в клетках постоянно, но в норме их уровень настолько низкий, что организм самостоятельно их нейтрализует с помощью антиоксидантной системы. Окислительный стресс происходит в том случае, когда активных форм кислорода образуется слишком много и антиоксиданты не могут полностью их инактивировать. Такой дисбаланс может вызвать окислительное повреждение белков, липидов или ДНК.
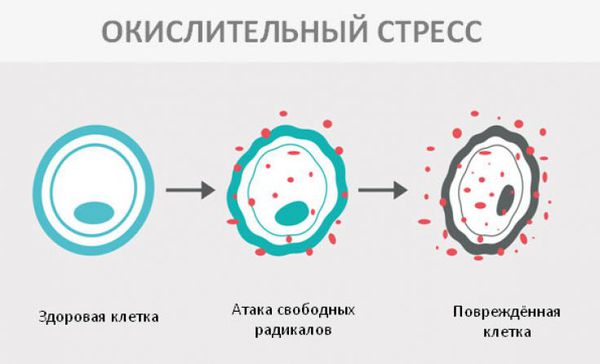
В ходе исследований окислительного повреждения макромолекул у пациентов с фенилкетонурией удалось установить, что увеличение концентрации фенилаланина в сыворотке крови (гиперфенилаланинемия) вызывает снижение антиоксидантоной защиты, что и приводит к окислительному стрессу [8] .
Ткани мозга особенно уязвимы для окислительного стресса из-за высокого потребления кислорода, высоких концентраций железа в тканях, низкого уровня антиоксидантной защиты, что способствует избыточному синтезу перекиси водорода (одной из форм активного кислорода) [7] .
Классификация и стадии развития фенилкетонурии
Согласно Европейской классификации, различают несколько вариантов течения данного заболевания [12] .
- Классическая фенилкетонурия характеризуется полным или практически полным дефицитом фенилаланин-гидроксилазы в печени. Сопровождается диетической переносимостью фенилаланина < 250-350 мг/день и концентрацией фенилаланина в крови при нормальной диете свыше 1200 мкмоль/л. При отсутствии своевременной диагностики первые клинические признаки заболевания отмечаются уже на первом году жизни ребёнка: родители жалуются на общую вялость, частые беспокойства, срыгивания при кормлении, проявления атопического дерматита. При осмотре ребёнка отмечается мышечная гипотония, задержка нервно-психического развития, в некоторых случаях — микроцефалия. Отличительной особенностью являются эпилептические приступы, плохо поддающиеся купированию стандартными схемами антиконвульсантной терапии.
- Умеренная фенилкетонурия характеризуется низкой остаточной ферментативной активностью (низкой способностью имеющихся ферментов участвовать в расщеплении фенилаланина и его превращении в тирозин), переносимостью фенилаланина от 350 до 400 мг/день и концентрацией фенилаланина в крови 600-1200 мкмоль/л. Данная форма отличается от классической фенилкетонурии более поздней манифестацией (чаще — первый-второй год жизни) и более медленным прогрессированием клинических признаков при отсутствии лечения.
- Лёгкая гиперфенилаланинемия. Уровень фенилаланина в плазме крови составляет менее 600 мкмоль/л при обычной диете. При данной форме у детей клинические признаки заболевания отсутствуют, либо незначительны. Несмотря на это, такие дети требуют особого контроля содержания фенилаланина в крови и непрерывной оценки нервно-психического развития.
Осложнения фенилкетонурии
Самым грозным осложнением фенилкетонурии является прогрессирующая умственная отсталость при отсутствии лечения. Однако данное заболевание ассоциируется также с рядом других патологий, влияющих на жизнь и здоровье пациента.
Доказано, что уровень депрессии среди взрослых с фенилкетонурией значительно выше и составляет около 18 % [10] . У них отмечается резкое снижение продуктивности, нарушения сна, выраженная утомляемость на фоне привычной деятельности, подавленное настроение, потеря интереса к жизни и привычным делам, нарушения аппетита, мысли о самоубийстве.
Также у взрослых пациентов с фенилкетонурией очень высока распространённость неврологических симптомов: тремор рук (присутствует у трети пациентов), генерализованная дистония (неритмичные медленные насильственные движения в различных частях тела, которые сопровождаются своеобразным изменением мышечного тонуса), задержка когнитивного и моторного развития (особенно при нарушениях диетотерапии), паркинсонизм [11] .
Пожизненная диетотерапия ассоциирована с нарушением роста, снижением минеральной плотностей костей и дефицитом питательных веществ, что требует постоянного контроля у профильных специалистов.
Диагностика фенилкетонурии
Первым эффективным тестом на определение у пациента гиперфенилаланинемии был бактериальный анализ ингибирования, разработанный американским микробиологом Робертом Гатри в 1963 году. Этот тест был основан на потребностях фенилаланина для роста культуры грамположительной бактерии Bacillus subtilis. Данный тест был очень удобен для массового скрининга, т. к. пятно засохшей крови можно было получить в кабинете врача, используя специальную фильтровальную бумагу, а затем отправить по почте в конверте в необходимую лабораторию [13] .
Развитие медицины привело к созданию тандемной масс-спектрометрии для быстрого определения концентраций аминокислот в небольших объёмах крови или плазмы. Данный метод даёт более низкую частоту ложноположительных результатов, измеряя уровни фенилаланина и тирозина в исследуемых образцах.
Основным методом ранней диагностики фенилкетонурии во всех странах мира принято считать неонатальный скрининг, который проводится в строго декретированные сроки для обеспечения своевременного начала лечения [12] . Неонатальный скрининг — это генетическое тестирование, которое позволяет выявить наиболее распространённые врождённые и наследственные заболевания у новорождённых детей.
В нашей стране проводится массовое обследование новорождённых детей на 5 наследственных заболеваний, одним из которых является ФКУ. Неонатальный скрининг проводится на четвёртые сутки жизни у доношенного ребёнка и на седьмые сутки у недоношенного ребёнка. В паспорте новорождённого и его истории болезни ставится пометка о проведении [14] .
Для неонатального скрининга медицинским персоналом с помощью скарификатора осуществляется забор крови из пятки новорождённого строго через 3 часа после кормления. Полученные образцы крови помещаются на специальные фильтровальные бумажные тест-бланки и отправляются в лабораторию.
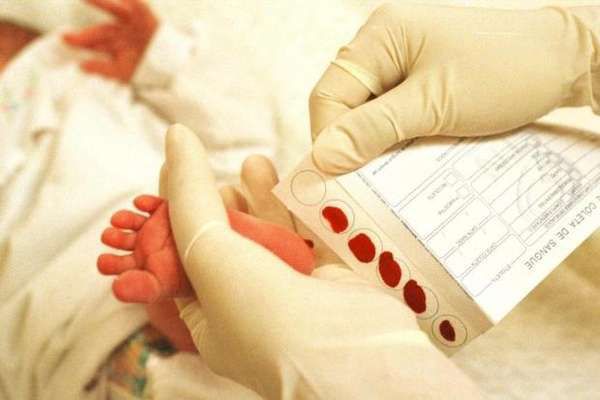
Стоит отметить, что у некоторых детей, особенно у рождённых раньше срока, может наблюдаться незрелость ферментных систем, участвующих в метаболизме аминокислот. Это приводит к кратковременному повышению фенилаланина и положительному результату при скрининге [12] Около 2 % всех случаев повышенного уровня фенилаланина в крови, выявленного при скрининге новорождённых, связаны с нарушением метаболизма кофермента BH4, который участвует в преобразовании фенилаланина. Это подчёркивает важность проведения дифференциальной диагностики при всех выявленных уровнях гиперфенилаланинемии. Фенилкеторурию необходимо дифференцировать с такими заболеваниями, как транзиторная гиперфенилаланинемия недоношенных, наследственная доброкачественная гиперфенилаланинемия, тирозинемия, галактоземия.
Генотипирование не является необходимым методом для диагностики фенилкетонурии, но помогает специалистам определять степень дисфункции белка, остаточную активность фенилаланин-гидроксилазы , и, следовательно, метаболический фенотип, на основании которого в дальнейшем будет строиться тактика лечения.
Лечение фенилкетонурии
В 1990 г. исследователь Smith и его коллеги в своей работе доказали, что отсрочка старта лечения фенилкетонурии на каждые четыре недели приводит к падению показателя коэффициента интеллекта (IQ) примерно на 4 балла [15] .
Последнее Европейское руководство по фенилкетонурии акцентирует внимание на том, что лечение заболевания следует начинать до достижения возраста 10 дней, что для многих стран потребует изменения сроков проведения неонатальных скринингов и ассоциированных с ним процедур. В нашей стране сроки проведения неонатального скрининга соответствуют рекомендациям Европейского руководства по ФКУ (4 сутки жизни у доношенных новорождённых и 7 сутки жизни у недоношенных) [16] .
Тактика контроля фенилкетонурии определяется, в первую очередь, уровнем фенилаланина в крови пациента. Среди специалистов существует единодушное мнение, что уровень фенилаланина более 600 мкмоль/л является прямым показанием к незамедлительному лечению. Кроме нейропедиатра Росы Гассио Субиракс и её соавторов, никто не описывал крупных исследований по изучению необходимости лечения пациентов при уровне фенилаланина менее 360 мкмоль/л [17] . Таким пациентам рекомендовано с особой тщательностью проводить мониторинг данного показателя в течение первого года жизни, так как с возрастом есть риск повышения уровня фенилаланина.
Контроль уровня фенилаланина в крови
Мнение медицинского сообщества относительно начала лечения пациентов с концентрациями фенилаланина 360-600 мкмоль/л достаточно противоречивы. Costello и соавторы [18] проводили исследование, в котором пациенты были разделены на три группы:
- 1 группа — концентрация фенилаланина < 400 мкмоль/л;
- 2 группа — концентрация фенилаланина 400-500 мкмоль/л;
- 3 группа – концентрация фенилаланина > 500 мкмоль/л.
Исследователи обнаружили тенденцию к снижению IQ у лиц с более высоким уровнем фенилаланина и рекомендовали лечение, которое бы обеспечило поддержку уровня фенилаланина в крови < 400 мкмоль/л в течение всего периода детства.
Согласно Российским Федеральным клиническим рекомендациям, при концентрациях фенилаланина более 360 мкмоль/л пациенту назначается лечение.
Диета при фенилкетонурии
На сегодняшний день основным доказанным методом лечения фенилкетонурии считается диетотерапия, которая основывается на трёх подходах:
- Ограничение естественного белка с использованием индивидуальной толерантности к фенилаланину.
- Использование добавок L-аминокислот (не содержащих фенилаланин) для перекрытия физиологических потребностей в микроэлементах.
- Употребление пищи с низким содержанием белка для удовлетворения физиологических потребностей пациента в энергии [19] .
Общее потребление белка должно обеспечивать безопасные уровни потребностей данного макронутриента с дополнительной дотацией 40 % L-аминокислот (20 % L-аминокислот необходимы для компенсации потребностей в незаменимой аминокислоте и еще 20 % L-аминокислот используются для контроля фенилаланина в крови).
При введении неадекватных дозировок L-аминокислот они ограничивают синтез белка. Белковый обмен, который в норме складывается из процессов анаболизма (синтеза) и катаболизма (распада) белков, смещается в сторону катаболизма. При данном процессе фенилаланин остается неиспользованным для синтеза белка, и его концентрация в крови будет расти.
При фенилкетонурии необходимо избегать продуктов, богатых белком (мясо, рыба, яйца, обычный хлеб, большинство сыров, орехи и семена), а также продуктов, содержащих аспартам (подсластитель, который используется при изготовлении некоторых газированных изделий и конфет). Пища с низким содержанием белка при диетотерапии данного заболевания должна содержать 50 мг или менее фенилаланина на 100 г сухого продукта. Фрукты и овощи, содержащие менее 75 мг фенилаланина на 100 г пищевого продукта, также могут быть включены в рацион. При употреблении таких овощей, как картофель, брокколи, цветная капуста, брюссельская капуста, нужно учитывать, что даже их небольшое количество в рационе обеспечивает организм 50 мг фенилаланина, поэтому их потребление не должно быть бесконтрольным. Для удобства родителей и ребёнка следует пользоваться «пищевым светофором», где все продукты разделены на группы, разрешённые (ограниченно или неограниченно) и запрещённые к употреблению.
Когда ребёнок маленький, соблюдение диеты не является проблемой для семьи, так как родители контролируют потребление продуктов. В младенческом возрасте предлагается использовать специализированные смеси без добавок фенилаланина, которые дополняются либо грудным молоком, либо стандартной семью для перекрытия суточной потребности в фенилаланине.
Дети более старшего возраста продолжают не только пить специальный безфенилаланиновый продукт, который способен обеспечить потребности в белках и калориях, но и получают дополнительное количество разрешённых продуктов (овощи и фрукты, мёд, животные и растительные масла, зефир, пастила, варенье), необходимых для создания пищевого разнообразия.
По мере взросления ребёнка соблюдение диеты становится всё труднее, так как дети с фенилкетонурией, в отличие от своих сверстников, значительно ограничены в выборе продуктов, что часто приводит к скачкам концентрации фенилаланина у подростков [20] [21] Долгосрочное поддержание диеты необходимо, поскольку пациенты после периода нарушений диеты намного труднее возвращаются к прежнему режиму питания.
Прогноз. Профилактика
Хороший прогноз для интеллекта возможен в том случае, когда пациенты с первого месяца жизни получают диету с пониженным содержанием фенилаланина. Существует пропорциональная связь между гиперфенилаланинемией и IQ у пациентов с фенилкетонурией, где при повышении данной аминокислоты на каждые 100 мкмоль/л происходит снижение IQ на 1,3-4,1 пункта [15] .
Если пациенты с фенилкетонурией получают и соблюдают лечение с самого раннего возраста, то их качество и продолжительность жизни ничем не отличается от их здоровых сверстников.
Пациенты, не получающие или не соблюдающие лечение, часто имеют инвалидность и низкий уровень качества жизни. Кроме того, несоблюдение диеты и отсутствие контроля фенилаланина в организме часто приводит к снижению продуктивности и внимания, нарушению поведения (особенно при уровне аминокислоты свыше 360 мкмоль/л).
Адекватное наблюдение за концентрацией фенилаланина в пределах допустимых показателей достаточно эффективно в профилактике большинства нарушений центральной нервной системы. Большинство людей демонстрируют нормальное общее развитие, легко справляются с образовательными стандартами, ведут самостоятельную жизнь и получают работу, будучи взрослыми.
При соблюдении пациентом режимов диеты и дополнительной дотации минеральных веществ не отмечается увеличения риска таких осложнений, как остеопороз или частые переломы при отсутствии других заболеваний костно-мышечной системы и соединительной ткани.
Профилактика фенилкетонурии
Если у будущих родителей или их близких родственников выявлена фенилкетонурия, то при планировании беременности рекомендуется проконсультироваться с генетиком и пройти обследование. Так можно определить риск рождения ребёнка с фенилкетонурией.
Чтобы предотвратить врождённые нарушения у ребёнка, женщинам с фенилкетонурией во время беременности следует придерживаться диеты с низким содержанием фенилаланина [22] .
Список литературы
- Loeber J.G. Neonatal screening in Europe; the situation in 2004 // J Inherit Metab Dis. — 2007; 30: 430-438.ссылка
- Guldberg P., Henriksen K.F., Sipila I., Guttler F., de la Chapelle A. Phenylketonuria in a low incidence population: molecular characterisation of mutations in Finland // J Med Genet. — 1995; 32: 976-978.ссылка
- Williams R.A., Mamotte C.D., Burnett J.R. Phenylketonuria: an inborn error of phenylalanine metabolism // Clin Biochem Rev. — 2008; 29(1): 31-41.ссылка
- Sumaily K.M., Mujamammi A.H. Phenylketonuria: A new look at an old topic, advances in laboratory diagnosis, and therapeutic strategies // Int J Health Sci (Qassim). — 2017; 11(5): 63-70.ссылка
- Surtees R., Blau N. Neurochemistry of phenylketonuria // Eur J Pediatr. — 2000; 159: 109–113. ссылка
- Velema M.,Boot E.,Engelen M., Hollak C. Parkinsonism in phenylketonuria: a consequence of dopamine depletion? // JIMD Rep. — 20: 35-38ссылка
- Halliwell B., Gutteridge J.M.C. Free radicals in biology and medicine. — New York: Oxford University Press Inc, 2007.
- Sitta A., Manfredini V., Biasi L., Treméa R., Schwartz I.V., Wajner M., et al. Evidence that DNA damage is associated to phenylalanine blood levels in leukocytes from phenylketonuric patients // Mutat Res. — 2005; 679:13-6. ссылка
- Yuwiler A., Geller E., Slater G.G. On the mechanism of the brain serotonin depletion in experimental phenylketonuria // J Biol Chem. — 1965; 240: 1170-1174.
- Ford S., O’Driscoll M., MacDonald A. Living with Phenylketonuria: lessons from the PKU community // Mol Genet Metab Rep. — 2018; 17: 57-63.ссылка
- Hofman D.L., Champ C.L., Lawton C.L., Henderson M., Dye L. A systematic review of cognitive functioning in early treated adults with phenylketonuria // Orphanet J Rare Dis. — 2018; 13(1): 150. ссылка
- van Rijt W.J., et al. Inborn Errors of Metabolism That Cause Sudden Infant Death: A Systematic Review with Implications for Population Neonatal Screening Programmes // Neonatology. — 2016; 109: 297–302. ссылка
- Ramagopalan S.V., Rakyan V.K. The promise and challenges of blood spot methylomics // Epigenetics. — 2013; 8(8): 775–777.ссылка
- Фенилкетонурия и нарушения обмена тетрагидробиоптерина. Клинические рекомендации. — М., 2016; 45.
- Smith I., Beasley M.G., Ades A.E. Intelligence and quality of dietary treatment in phenylketonuria // Arch Dis Child. — 1990; 65(5): 472–478.ссылка
- van Wegberg A.M.J., MacDonald A., Ahring K., et al. The complete European guidelines on phenylketonuria: diagnosis and treatment // Orphanet J Rare Dis. — 2017; 12(1): 162. ссылка
- Gassio R., Artuch R., Vilaseca M.A., Fuste E., Boix C., Sans A., et al. Cognitive functions in classic phenylketonuria and mild hyperphenylalaninaemia: experience in a paediatric population // Dev Med Child Neurol. — 2005; 47(7): 443–448.ссылка
- Costello P.M., Beasley M.G., Tillotson S.L., Smith I. Intelligence in mild atypical phenylketonuria // Eur J Pediatr. — 1994; 153(4): 260–263.ссылка
- MacLeod E.L., Gleason S.T., van Calcar S.C., Ney D.M. Reassessment of phenylalanine tolerance in adults with phenylketonuria is needed as body mass changes // Mol Genet Metab. — 2009; 98: 331–337.ссылка
- Walter J.H., White F.J. Blood phenylalanine control in adolescents with phenylketonuria // Int J Adolesc Med Health. — 2004; 16: 41–45.ссылка
- Crone M.R., van Spronsen F.J., Oudshoorn K., Bekhof J., van Rijn G., Verkerk P.H. Behavioural factors related to metabolic control in patients with phenylketonuria // J Inherit Metab Dis — 2005; 28: 627-37.ссылка
- Mayo Clinic. Phenylketonuria (PKU). — 2018.
Фенилкетонурия
Фенилкетонурия (ФКУ) ― наследственное нарушение метаболизма аминокислот, в первую очередь фенилаланина (ФА), входящего в состав белков. Вещество участвует в укладке белка и стабилизации белковых структур.
Медицинские услуги
Анализы
Первые симптомы: частое срыгивание, рвота, экземы, судороги, исходящий от мочи и кожи запах плесени. Ребенок может был вялым либо, наоборот, гиперактивным. Отстает в психомоторном развитии, наблюдаются признаки олигофрении. Диагноз может быть поставлен в родильном доме. Все дети с фенилкетонурией безусловно получают статус «ребенок-инвалид».
Лечение заболевания заключено в соблюдении специальной низкобелковой диете, не содержащей продукты с ФА.
Определение заболевания
Фенилкетонурия ― это врожденная, генетическая патология, подразумевающая нарушения гидроксилирования фенилаланина. Характеризуется накоплением в организме аминокислоты и продуктов ее метаболизма, что ведет к тяжелым поражениям центральной нервной системы. Впервые заболевание было описано норвежским врачом И. А. Феллингом в 1934 году.
Изучая болезнь специалисты установили, что за наличие болезни отвечает единственный ген фенилаланингидроксилазы. Первое успешное лечение было разработано и проведено в 1950 году в Англии.

В неонатальном периоде клиника отсутствует. Патология проявляется в первые полгода жизни ребенка. В дальнейшем накопление вещества приводит к тяжелым нарушениям развития. Поэтому крайне важно сразу после рождения выявить дефект и не допустить употребление продуктов, содержащих фенилаланин. Более позднее соблюдение диеты не устранит полученные нарушения, но не допустит развития новых.
Патология одинаково часто встречается среди лиц обоих полов. Расовых особенностей не выявлено. Большое количество больных в таких странах как Китай, Турция, Ирландия. В среднем по России с фенилкетонурией рождается каждый 7-ми тысячный ребенок.
Причины фенилкетонурии
Существует три типа генетического отклонения, первый считается классическим, поскольку диагностируется в более чем 90% случаев. Второй и третий ― более редкая форма патологии. Симптоматика схожа во всех типах, заболевание приводит к умственной отсталости. При классической форме фенилкетонурии избежать этого можно диетотерапией, но атипичные варианты, к сожалению, коррекции не подлежат.
- Классическая фенилкетонурия (I тип) ― это низкая выработка фенилаланингидроксилазы (ФАГ), что приводит к собиранию в естественных жидкостях организма фенилаланина и продуктов его расщепления. Патология вызвана мутированным геном РАН.
- Фенилкетонурия II типа ― недостаток дигидроптеридинредуктазы, что препятствует преобразованию фенилаланина в тирозин. Патология из-за мутации гена QDPR.
- Фенилкетонурия III типа ― недостаток 6-пирувоилтетрагидроптеринсинтазы, нужной для синтеза тетрагидробиоптерина. Патология вызвана мутированным PTS-геном.
Все формы заболевания наследуются по аутосомно-рецессивной форме. Это означает, что генетический дефект может быть унаследован у одного из родителей. Половая принадлежность родителя и ребенка не играет роли.

Классификация
Фенилкетонурия в настоящее время не имеет общепризнанной мировой классификации. Над этим вопросом ведутся дебаты, наравне с изучением заболевания. Чуть ранее, до расшифровки генов, считалось, что степень поражения интеллектуальных способностей зависит от степени активности фермента. Поэтому текущая квалификация признана устаревшей. Не учитывает она и другие симптоматические факторы.
При диагностировании ставят:
- I тип (дефицит ФАГ) ― концентрация ФА больше 20 мг/дл.
- Средняя форма ФКУ ― ФА от 8,1 до 20 мг/дл.
- Легкая форма ГФА-уровень ― ФА от 2,1 до 8,0 мг/дл.
При уровне до 8,0 мг/дл фенилкетонурию считают доброкачественной. Она не требует специального лечения, но необходимо наблюдение первый год жизни ребенка. Контролирует состояние врач-педиатр, невролог, генетик.
Выделяют также еще одну форму фенилкетонурии, не требующую коррекции. Это транзиторная форма ГФА в период новорожденности. Возникает, как правило, при недоношенности, что обусловлено функциональной незрелостью организма. Транзиторная фенилкетонурия ― это временное повышение ФА-уровня, способное подняться до критических значений. При этом клиника отсутствует либо проявления весьма незначительны. Через несколько месяцев биохимические показатели приходя в норму.
Патогенез
Механизм зарождения и развития фенилкетонурии связан с нарушением обмена органического соединения ― аминокислоты фенилаланина. Метаболический блок препятствует преобразованию фенилаланина в тирозин. Аминокислота не только не преобразуется, а накапливается в виде токсичных метаболитов:
- фенилмолочная кислота;
- фенилпировиноградная кислота;
- фенилуксусная кислота;
- фенилэтиламин и прочее.
Скопление фенил-веществ оказывает токсическое действие на ЦНС. В настоящий момент механизм еще до конца не изучен, врачам не известен патогенез дисфункции головного мозга.
Существуют предположения, что поражение нервной системы является результатом ряда факторов. Среди них как прямое токсического воздействие фенилаланина, так и нарушение обмена белков, липопротеидов и гликопротеидов, сбой гормонального метаболизма и мембранного транспорта аминокислот. Все это в комплексе имеет важное значение для созревания и правильного функционирования ЦНС.
Симптомы
I тип. Первые признаки у ребенка проявляются в возрасте от 2 месяцев до полугода.
- Апатичность либо, наоборот, повышенная раздражительность.
- Отсутствие интереса к окружению, людям, предметам, обстановке.
- Частое срыгивание.
- Аллергический дерматит.
- Нарушение мышечного тонуса.
- Пониженное давление.
- Судороги.
- Иногда развивается микроцефалия (малый размер черепа относительно других частей тела) и гидроцефалия (избыточная жидкость, омывающая головной мозг).
К характерным симптомам относятся гипопигментация кожи, волос, радужной оболочки глаз. Моча имеет специфический запах плесени или его еще называют «мышиным» запахом. Эпилептические припадки наблюдаются у половины больных, часто является первым выраженным клиническим симптомом. Приступ характеризуется «салаамовыми» судорогами, напоминающими кивки. Они случаются часто, плохо поддаются антиконвульсантному лечению.
Если не корректировать концентрацию ФА, болезнь прогрессирует. Как правило, уровень IQ у таких детей не превышает 20, при норме от 85. Умственная отсталость настолько сильная, что отсутствуют эмоциональные реакции, наблюдаются психопатии и шизофреноподобные расстройства.
II тип. Первая симптоматика проявляется на первом году жизни.
- Повышенная возбудимость.
- Задержка развития.
- Обильное слюнотечение.
- Сниженное артериальное давление.
- Частое повышение температуры тела.
- Сухожильная гиперрефлексия (повышение рефлексов) или спастический тетрапарез (обессиливание всех четырех конечностей).
- Миоклоническая эпилепсия (генерализованные приступы, преимущественно возникающие после пробуждения).
- Микроцефалия.
Отличительная особенность второго типа ― гибель нейронов, нарушение метаболизма фолатов, а также кальцификация в различных отделах головного мозга. Болезнь быстро прогрессирует, может привести к смерти ребенка в течение 2 — 3 лет.
III тип. Симптомы дефицита пирувоилтетрагидроптеринсинтетазы схож с проявлениями болезни Паркинсона:
- Постуральная нестабильность и трудности походки. Сложно либо невозможно поддерживать определенное положение всего тела или конечностей.
- Гипокинезия (низкая двигательная активность, ограниченный темп и объем движений).
- Гиперсаливация (повышенное слюноотделение).
- Нарушения глотания.
- Окулогирные кризы (симметричное отклонение обоих глаз, обычно направленное вверх).
В 80% случаев этот тип заболевания сопровождается снижением количества биогенных аминов в ликворе. Лечение затруднено тем, что раннее снижение концентрации ФА может вызвать серьезные патологические изменения. Несоблюдение диетотерапии приведет к замедлению развития речи, низкому интеллекту, проблемам с памятью.
Диагностика
Выявить фенилкетонурию можно в первые дни после рождения до появления какой-либо симптоматики. Для определение концентрации фениламина в крови проводят:
- микробиологический тест;
- хроматографию;
- флюориметрию;
- масс-спектрометрию.
Во всех случаях биологическим материалом выступают сухие пятна капиллярной крови младенца.
С недавнего времени анализ на фенилкетонурию входит в программу неонатального скрининга. Его проводят всем новорожденным, особенно важно исследование для недоношенных детей. Критерий диагностирования ― повышенная концентрация фенилаланина при норме 0 — 2 мг/дл. Повышенный уровень требует проведения уточняющей диагностики. Потребуется установить сам факт наличия фенилкетонурии и выявить ее причину.
Если скрининг-тест показал высокие результаты уровня ФА, дополнительно может быть назначено:
- Фенилаланин-нагрузочная диагностика для выявления нозологической формы заболевания.
- Молекулярно-генетический анализ для установления формы: классическая, II или III тип.
- Секвенирование гена РАН, если молекулярно-генетическая диагностика дала отрицательный результат по гену ФАГ.
- Анализ на птерины в урине для исключения птерин-зависимых форм.
Дифференциальное диагностирование фенилкетонурии проводят с такими патологиями, как нарушение функции печени, галактоземия и с другими заболеваниями.
Лечение
Симптоматическая терапия при любой формой фенилкетонурии неэффективна. Существует только один способ предотвратить негативные последствия заболевания ― диетотерапия. Из рациона исключают высокобелковые и содержащие фенилаланин продукты. Недостающее количество белка восполняют специализированным лечебным питанием, с максимально низким содержанием аминокислоты ФА или полностью ее лишенным. Следует учитывать, что эффективность терапии напрямую зависит от времени начала коррекции и уже произошедших патологических изменений.
Цель лечебного питания при классической форме заболевания ― это предотвращение развития нарушений ЦНС, физического и умственного развития. Легкая форма ГФА допускает расширение диеты под строгим наблюдением врача за состоянием ребенка и биохимическими показателями. Под запретом: мясо, рыба, орехи, шоколад и бобовые, все виды яиц, творог и сыры. Также следует исключить продукты, содержащие искусственный подсластитель аспартам.
Критерий эффективности лечения ― уровень ФА в крови.
Прогноз и профилактика
Проведения массового скрининга в родильных домах позволяет своевременно выявить генетическое отклонение. Вовремя начать соблюдение диетотерапии и, как следствие, предотвратить тяжелые последствия. В противном случае прогноз в отношении умственного развития неблагоприятный.
Классическая ФКУ имеет благоприятный прогноз если диагностирована в первые недели жизни ребенка и соблюдаются все требования врачей. Такие дети ходят в обычные школы, способны получить высшее образовании и вести нормальный образ жизни.

Во время подготовки к беременности пара должна пройти предварительное генетическое тестирование на наличие мутаций в гене РАН. Если у одного из родителей есть дефектный ген, шанс родить ребенка с ФКУ 1:4 и 100% если оба родителя больны.
Женщины с установленной фенилкетонурией при беременности и грудном вскармливании должны соблюдать строгую диету. Высокая концентрация аминокислоты в крови и околоплодных водах оказывает серьезное тератогенное воздействие на плод.
Преимущества АО «СЗЦДМ»
Сдать анализ на уровень ФА можно в подразделениях АО «СЗЦДМ» Здесь вас ждет:
- квалифицированных и доброжелательный персонал;
- новейшее оборудование, отправка результатов исследований по эл. почте;
- несколько вариантов получения данных анализов;
- удобное расположение терминалов;
- отсутствие очередей, условия конфиденциальности.
Лаборатории находятся в Санкт-Петербурге и других города Ленинградской области, а также в Великом Новгороде, Новгородской обл., Пскове, Калининграде.
Обратите внимание!
Данный материал подготовлен исключительно в информационных целях. Не используйте информацию для самодиагностики и самолечения, поскольку каждый случай индивидуален и оценивать ситуацию должен профильный специалист.
Не рассматривайте данную статью как альтернативу консультации с врачом! Выберите своего врача по ссылке
Источник https://muzoktcrb.ru/blog/fenilketonuriya.html
Источник https://probolezny.ru/fenilketonuriya/
Источник https://cdmed.ru/diseases/genetika/fenilketonuriya/