Фенилкетонурия
Фенилкетонурия (либо ФКУ) — это наследственное, редко встречающееся заболевание, которое проявляется в нарушении обмена аминокислот. Если точнее, то организм людей с такой болезнью не может расщеплять фенилаланин, поступающий вместе с белковой пищей. Из-за такого дисбаланса в теле накапливаются опасные соединения, способные отравлять нервную систему, в том числе и ткани головного мозга. Эти отравления могут быть настолько сильными, что у ребенка развивается умственная отсталость, включая идиотию.
Несмотря на серьезную опасность болезни, сейчас ее вполне можно нейтрализовать — то есть дети, которые рождаются с такой проблемой, не обречены.
Статистика
Количество детей с таким заболеванием зависит от страны, местности проживания. Так, в России фиксируют рождение одного малыша, страдающего фенилкетонурией, на 10 000 человек. В Великобритании этот показатель в два раза выше, а у африканских детей подобная проблема почти не встречается. Девочки страдают этим недугом чаще — почти в два раза по сравнению с мальчиками.
Симптомы
У детей с ФКУ при рождении нет характерных симптомов, которые позволят сразу понять, что с малышом что-то не так. Ребенок с такой проблемой выглядит абсолютно здоровым. И если такой малыш употребляет в пищу белковую еду с большим количеством фенилаланина, начинаются первые симптомы. По этой причине диагностика проводится массово, всем детям сразу после рождения (см. ниже).
Если по какой-то причине диагностика не была проведена, и ребенок с болезнью получает белок, со временем у него начнутся тревожные симптомы. Первыми будут слабость, беспокойство, отсутствие у малыша улыбки, ярких реакций на окружающий мир. Дальше идут симптомы медленного развития: например, когда приходит время, ребенок не пытается сесть, перестает узнавать маму и т. д.

По мере развития болезни могут проявляться такие симптомы фенилкетонурии:
- Изменение цвета волос и глаз к более светлым. Это связано с тем, что в организме недостаточно меланина.
- Прибавление веса — малыш начинает быстро поправляться.
- Высыпания, экзема, сухость и шелушение кожи.
- Частая рвота.
- Неприятный («мышиный») запах мочи
К двум-трем годам основные диагностические признаки фенилкетонурии усиливаются и выглядят уже так:
- Спазмы и судороги, тремор пальцев рук.
- Постоянно зажатая поза. Ребенок не может расслабиться из-за сильного напряжения в мышцах.
- Неадекватные действия. Так, малыш может резко засмеяться, закричать — то есть просто ведет себя неадекватно ситуации.
- Деформация ушей, уменьшение черепа в отношении размеров тела.
- Выступающая нижняя челюсть.
- Недержание.
На фоне заболевания у ребенка развиваются серьезные и устойчивые психические отклонения, которые уже невозможно исправить. В результате пациент становится инвалидом. Но, подчеркнем еще раз: так развиваются события только в том случае, если не была проведена своевременная диагностика заболевания.
Причины
Поскольку речь идет о генетическом заболевании, причины фенилкетонурии сводятся исключительно к мутации гена 12-й хромосомы. Именно этот ген отвечает за определенный фермент, помогающий перерабатывать аминокислоту фенилаланин. Из-за мутации количество фермента становится меньше, поэтому сама аминокислота накапливается в тканях, отравляя их. Все это влияет и на нейромедиаторы, работу нервных проводников.
Заболевание возникает тогда, когда патологические гены матери и отца совпадают, оно не зависит от пола ребенка.
Названная причина фенилкетонурии определяет развитие болезни в 98% случаев, однако есть еще 2%, когда проблема заключается в других генетических дефектах. Если в первом случае (почти всегда) назначается лечение диетой — и оно очень эффективно, то во второй ситуации оно будет бесполезным.
Диагностика
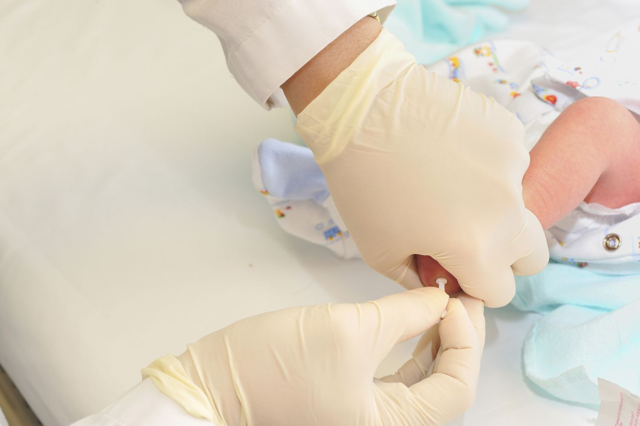
Определить риски появления этого заболевания можно еще до зачатия ребенка — если оба родителя пройдут генетическую экспертизу. Но после рождения малыша его обязательно обследуют на наличие фенилкетонурии. В России это обязательная процедура, которая входит в неонатальный скрининг. Анализ обязательно нужно взять в течение первых трех недель жизни, так что, если малыш появился на свет не в роддоме, родителям обязательно нужно об этом позаботиться. Результаты анализа можно получить через сутки.
Если в анализе обнаружен ген с изменениями, то малыша и родителей обследуют в медико-генетическом центре. Есть ситуации, когда диагноз после тщательного изучения всех данных опровергается. Но чтобы подтвердить его или убрать, необходимо провести дополнительную диагностику фенилкетонурии:
- изучить сыворотку и сухое пятно крови,
- провести потовый тест,
- сделать копрограмму,
- провести ДНК-диагностику.
Затягивать с рекомендациями врачей нельзя ни в коем случае. Занимаются лечением педиатры и эндокринологи.
Лечение
Основное лечение фенилкетонурии сводится к тому, чтобы убрать из рациона ребенка животные белки. Если это будет сделано в первые недели жизни, малыш будет развиваться нормально — и умственная отсталость ему не грозит. Чем позже будет пересмотрен рацион, тем выше риски, что процесс будет запущен, а потому его можно будет только затормозить.
Строгая диета соблюдается больными в среднем до 18 лет — после этого возраста белки допускаются, но их количество необходимо контролировать. Если в последующем девочка с таким диагнозом вырастет и захочет родить, до зачатия, а также во время беременности и лактации ей нужно будет вернуться к строгой диете без белка. Чтобы восполнить питательные вещества, которые ребенок не может получить из пищи, ему нужно заменить белковую еду специальными продуктами с пептидами и свободными аминокислотами. Для этого есть целенаправленно разработанные решения, с которыми родителей ознакомят врачи. Также при лечении фенилкетонурии можно кормить малышей грудным молоком, но маме придется тоже следить за питанием и сидеть на специальной диете.
Диета ребенка зависит и от возраста. Если сначала важно исключить животный белок, то в дошкольном и школьном возрасте из рациона убирается любой белок. Строго необходимо соблюдать и возрастные нормы фенилаланина.
Поскольку основные диагностические признаки фенилкетонурии нельзя определить визуально, а только по анализам крови, детям с таким диагнозом нужно регулярно сдавать кровь и контролировать количество фенилаланина. Нормой в данном случае являются показатели 180-240 мкмоль/л либо 3-4 мг%. Регулярность анализов также зависит от возраста. До трех месяцев ребенка проверяют каждую неделю, затем постепенно количество визитов к врачу снижается.
Если у вашего ребенка выявлена классическая фенилкетонурия, и вы хотите получить консультацию опытного эндокринолога, а также сдать очередные анализы, клиника АО «Медицина» в Москве — место, где вам обязательно помогут. Большой опыт лечения детей с самыми разными недугами позволяет профессионалам подбирать оптимальные программы восстановления и поддержания для каждого малыша.
 Профилактика
Профилактика
Поскольку речь идет о генетическом заболевании, специфической профилактики фенилкетонурии просто не существует. Родители могут сдать специальный анализ до зачатия и посмотреть, каковы риски, что у них родится ребенок с таким недугом. Но независимо от того, был ли пройден генетический тест, всех новорожденных малышей в роддоме все равно проверяют.
Единственная профилактическая мера касается тех родителей, которые по каким-то причинам приняли решение рожать в домашних условиях или в странах, где проверка на фенилкетонурию не проводится обязательно. Им необходимо в течение первых трех недель жизни малыша сдать анализы крови и проверить новорожденного на наличие искаженного 12-го гена.
Вопросы и ответы
Как влияет фенилкетонурия на умственное развитие ребенка?
Если вовремя сесть на диету (с первых недель жизни малыша), то заболевание никак не повлияет на развитие. При грамотном лечении ребенок вырастет полноценным и сможет учиться, социализироваться — то есть не почувствует на себе никаких ограничений кроме тех, что связаны с диетой.
Что будет, если начать диету значительно позже?
К сожалению, задержка в развитии в этом случае не компенсируется. В ряде случаев ее можно задержать — и ребенок будет жить достаточно комфортно. Но очень часто это лишь замедление патологических процессов, так что своевременное лечение просто необходимо.
Можно ли избежать фенилкетонурию?
Поскольку заболевание обусловлено генетически, избежать его, когда встречаются два «поломанных» гена, невозможно. Родители могут только предотвратить проблемы, вызванные болезнью. Но сделать так, чтобы малыш точно не заболел, на данном этапе развития медицины невозможно.
Фенилкетонурия


- Лечащие врачи: Генетик, Невролог, Терапевт, Эндокринолог
- Диеты при болезни: Безбелковая диета
Общие сведения
Фенилкетонурия относится к наследственным заболеваниям из группы ферментопатий. Она связана с нарушением обмена ароматических аминокислот, а конкретно — фенилаланина. Если человек с этим заболеванием не соблюдает низкобелковую диету, то в его организме происходит накопление фенилаланина и продуктов метаболизма, обладающих токсическим действием на ЦНС, которое в свою очередь вызывает поражения нервных структур и приводит к нарушениям умственного развития. Возникает фенилпировиноградная олигофрения по аутосомно-рецессивному типу наследования, однако на сегодняшний день это одно из немногих наследственных заболеваний, которое поддается успешному лечению.
Распространённость не зависит от гендерного различия, но отличается у разных групп населения. Однако, риску летального исхода более подвержены мальчики возрастом до 1 года.
В европеоидной расе и среди жителей Америки фенилкетонурия встречается у 1 человека на 10-15 тыс. Наиболее высокая частота случаев зарегистрирована у граждан Турции: 1 из 2,60 тыс. Финляндия и Япония отличается наиболее низкой частотой встречаемости данного недуга не более 1 человека на 120 тыс. рождений. Сенсацией были данные исследований 1987 г, когда среди представителей цыганских популяций в Словакии был обнаружен сверхвысокий уровень фенилкетонурии, вызванный предположительно инбридингом: болезнь выявляли у каждого 4-ого ребенка.
Историческая справка
В 1934 г норвежским врачом Иваром Асбьёрном Феллингом была выявлена гиперфенилаланинемия. Она оказалась ассоциирована с задержкой умственного развития. У жителей Норвегии заболевание называлось в честь его открывателя — болезнь Фёллинга.
Методика успешного лечения фенилкетонурии впервые была разработана в стенах Бирмингемского детского госпиталя (Англия). Ее разработал Хорст Биккель со своей командой медиков в начале пятидесятых годов двадцатого века. Однако, больший успех терапия имела, когда удалось широко применять раннюю диагностику недуга, вызванного повышенным содержанием фенилаланина в кровяном русле новорождённых по методу Гатри, разработанному и внедренному в 1958-1961 годах.
Фенилкетонурия, что это за заболевание?
Название происходит от двух названий химических соединений – фенилаланина и кетонов, а также греческого слова uron означающего – моча. Это заболевание вызывает расстройства движений и тонуса мышц, а также отставание физического развития и прогрессирующее слабоумие, поэтому еще называется фенилпировиноградной олигофренией.
На начальных этапах фенилкетонурия никак себя не проявляет и протекает бессимптомно. Тревожными знаками становится манифестация симптомов в виде сонливости и плохо аппетита у детей 6-12 месяцев. Такие дети отличаются от родственников и ровесников слишком светлой кожей, блондинистыми волосами и голубым цветом глаз. Также характерным симптомом становится развитие сыпи, напоминающей дерматит или экзему. Отсутствие лечения приводит к заметной, значительно выраженной задержке умственного развития, поэтому очень важно выявить болезнь на первых годах жизни, пока не успели произойти необратимые патологические изменения.
Патогенез
Как незаменимая аминокислота – фенилаланин является протеиногенной и играет важную роль в построении белковых молекул всех живых организмов, обеспечивая стабилизацию белковых структур и фолдинг (нативную укладку), являясь составным звеном функциональных центров. Необходим для синтеза гормонов щитовидной железы (в большей мере — тироксина), а также адреналина и меланина.
Метаболический блок превращения фенилаланина в тирозин приводит к его накоплению и активации побочных путей его обмена (например, печенью), в результате чего в тканях организма аккумулируются его токсичные производные — фенилпировиноградная, фениломолочная и другие кетоновые кислоты, в норме которых практически не должно образовываться. Помимо этого, происходит синтез ортофенилацетата и фенилэтиламина – соединений также почти полностью отсутствующих при нормальном метаболизме, причем производные последнего являются психоделиками и психостимуляторами. Наличие их в избытке инициирует нарушение обмена жиров в структурах головного мозга. Считается, что именно этот фактор вызывает прогрессирующее снижение интеллекта и может привести в самой глубокой степени умственной отсталости — идиотии. На сегодняшний день остаются до конца невыясненными механизмы развития нарушений работы мозга, вызванные фенилкетонурией. Предположительно, этому способствует дефицит нейромедиаторов (серотонина, дофамина) в синапсах мозга на фоне прямого токсического действия фенилаланина и относительного снижения количества тирозина и прочих «крупных» аминокислот, способных конкурировать с фенилаланином в процессах переноса через гематоэнцефалический барьер.
Гиперфенилаланинемия приводит к нарушениям обмена липо- и гликопротеидов, аминокислотного равновесия как жидкостных сред, так и клеточных структур. В основе клинических проявлений также лежат нарушения метаболизма гормонов, обмена моноаминовых нейромедиаторов таких как катехоламины и серотонин.
Разлад функции печени выражен в виде диспротеинемии, генерализованной гипераминоацидемии, метаболического ацидоза, нарушения белковосинтезирующей и окислительной деятельности клеточных органелл.
Классификация
Различают типичную (классическую) и атипичные формы (варинатные, коферментзависимые, злокачественные — на них приходится примерно 10% случаев) фенилкетонурии в связи с генетической и клинической неоднородностью аномалий обмена фенилаланина, а также кофактора — биоптерина.
Причиной второй диеторезистентной атипичной формы фенилкетонурии является дефицит дигидроптеридинредуктазы в результате генного дефекта локализующегося на участке 4р 15.3 хромосомы. Открыл ее Смит в 1974 году. Энзимный дефект нарушает процесс восстановление активной формы тетрагидробиоптерина, принимающего участие в виде кофактора при гидроксилировании фенилаланина в тирозин, а также предшественников нейромедиаторов серотонинового и катехоламинового ряда, таких как L-ДОФА и 5-окситриптофан.
Фенилкетонурию 3 диеторезистентную описал Кауфман в 1978 г. (согласно источнику Википедия). Она отличается недостаточностью 6-пирувоил тетрагидроптерин синтетазы, принимающей участие в синтезе тетрагидробиоптерина.
Атипичные формы отличаются прогрессированием проявлений заболевания.
Другие формы патологии связаны с нарушением иных альтернативных путей метаболизма фенилаланина с формированием метилминдальной ацидурии и парагидроскифенилуксусной ацидурии.
Причины возникновения фенилкетонурии
Благодаря накоплению опыта в области диагностики и лечения фенилкетонурии с течением времени стало понятно, что причины возникновения этого наследственного недуга кроются в мутациях гена, кодирующего фенилаланин-4-гидроксилазу — 12q23.2. Тип наследования аутосомно-рецессивный.
Чаще всего болезнь вызвана резко сниженной или полностью отсутствующей активностью такого фермента печени как фенилаланин-4-гидроксилаза, которая в норме необходима для катализации превращения (гидроксилирования) фенилаланина в аминокислоту — тирозин.
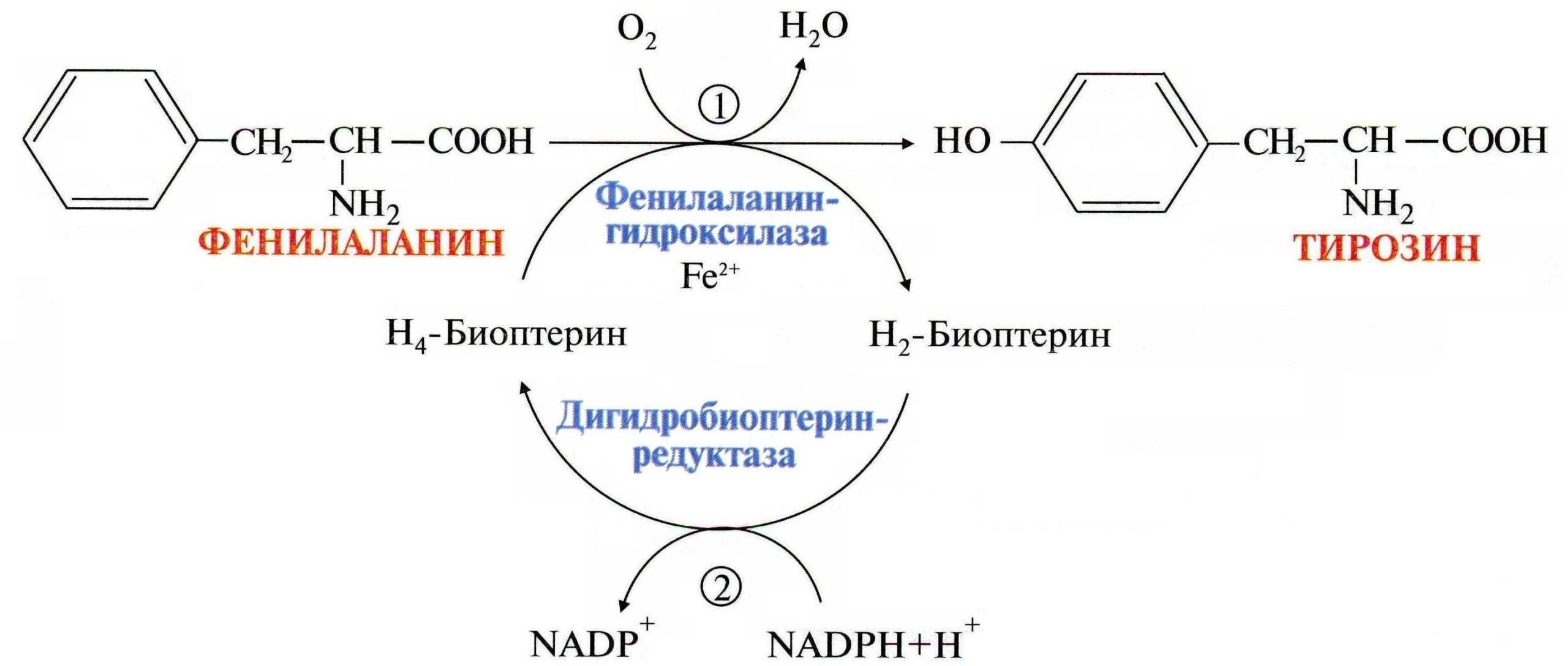
Обмен фенилаланина и тирозина
Однако, примерно в 10% случаев гиперфенилаланинемии воздействую атипичные факторы, связанные с мутациями в иных генах, кодирующих ферменты, обеспечивающие синтез кофактора фенилаланингидроксилазы, известного как тетрагидробиоптерин (BH4).
Симптомы фенилкетонурии
Фенилкетонурия отличается достаточно ярко выраженной клинической картиной, включающей такие симптомы как:
- отставание физического развития с 6 месячного возраста;
- микроцефалия;
- вегетативные дисфункции;
- повышенная возбудимость и двигательная гиперактивность;
- экзема или дерматит, возможно с папулезными кожными высыпаниями (как на фото ребенка с фенилукетонурией);
- мышечная гипертензия;
- атаксия;
- гиперкинезы;
- неустойчивость походки;
- судорожные припадки;
- нередко обнаруживаются пороки сердца;
- чувствительность к травматизации и осветление кожи, волос и радужки глаз (депигментация), в особенности при несоблюдении необходимой диеты, вызывающей недостаточность меланина в организме, являющегося производным тирозина.
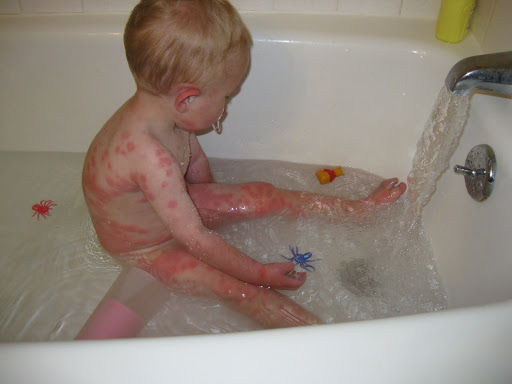
Экзема при фенилкетонурии
Психические нарушения
Фенилкетонурия вызывает значительные патологические изменения обменных процессов в головном мозге, что влечет за собой следующие нарушения:
- глубокая степень умственной отсталости, вплоть до идиотии или имбецильности;
- трудности в обучении;
- возникновение явлений эхопраксии — повторение движений за окружающими;
- эхолалии (повторение речи);
- вялое поведение может сменяться редкими вспышками злости и раздражительности.
Как можно увидеть на фото больных фенилкетонурией телосложение их обычно диспластическое, размеры черепа уменьшены, отмечается гипогенитализм и нанизм.
Анализы и диагностика
В основе диагностики фенилкетонурии лежат лабораторные анализы крови и мочи, изучают биоптаты печени, а также проводят генетические исследования:
- полуколичественный тест или количественное определение фениаланина в кровяном русле, в норме составляет 0,01-0,02 г/л, но при фенилкетонурии присутствует во всех жидких средах организма, а в сыворотке крови повышается до 0,15-0,2 г/л;
- на ранних этапах жизни (у детей 10-12 дней) могут быть выявлены в моче продукты распада фенилаланина – фенилкетоны, в особенности при нелеченой клинической картине, при этом используется проба Феллинга с 5-10% раствором FeCI3 или индикаторные бумажки «Фенистикс» и «Биофан», меняющие при положительном результате цвет на сине-зеленый;
- в печеночном биоптате исследуют уровень активности фенилаланингидроксилазы;
- проводят поиск мутаций в хромосомах гена фенилаланингидроксилазы, тогда как для 2 и 3 типа заболевания, связанного с мутациями в гене, отвечающем за биосинтез кофактора необходимо выполнение дополнительных диагностических исследований – выявления в моче низкого количества продуктов обмена нейротрансмиттеров, таких как гомованилиновая, ванилилминальная и оксииндолилуксусная кислоты.
На 3-4 сутки жизни новорожденного наиважнейшим в обнаружении обменных врождённых заболеваний оказывается неонатальный скрининг посредством анализов крови. Этот этап делает возможным обнаружить фенилкетонурию и как можно раньше начать лечение, чтобы предотвратить необратимые последствия. В РФ таким детям устанавливается категория инвалидности до 18 лет.
Лечение фенилкетонурии
Наиболее благоприятным оказывается прогноз при ранней диагностике заболевания. Если же недуг выявляется поздно, то практически невозможно справиться с уже резвившимися необратимыми изменениями тканей мозга.
В основе лечения фенилкетонурии – строгая диета, ограничивающая животные и растительные белки, которая должна длиться минимум до полового созревания, а может и пожизненно. Постоянно нужно быть в контакте с лечащим врачом, который будет мониторить состояния и психологически поддерживать пациента. При отмене диеты регулярно проводят психологические тесты, электроэнцефалограмму и определяют уровень фенилаланина в кровяном русле. Процесс ослабления диеты начинают примерно в 8-10 лет, когда заканчиваются процессы миелинизации мозга.
В случаях некоторых (мягких) форм заболевания возможно лечение кофактором (тетрагидробиоптерином) при поражённом ферменте — фенилаланингидроксилазе. Перспективными считаются разработки новых подходов лечения фенилкетонурии — использования заместительной терапии с применением фенилаланинлиазы (PAL, пегвалиазы) — растительного фермента, завершающего метаболизм фенилаланина безвредными метаболитами, а также генотерапия, в основе которой введение в организм вирусного вектора с геном фенилаланингидроксилазой. Такие методы не смогли пока выйти из стен лабораторных исследований.
При атипичных формах, не поддающихся диетотерапии, лечение сводится к введению препаратов тетрагидробиоптерина либо его синтетических аналогов, к примеру, сапроптерина.
Наиболее перспективной методикой лечения данного тяжелого заболевания является генотерапия как классический образец успешной борьбы и организационной помощи при наследственных патологиях.
Фенилкетонурия (ФКУ)
Фенилкетонурия (ФКУ) ― редкое заболевание, связанное с отсутствием способности усваивать аминокислоту фениланин. Поскольку она довольно распространена в рационе человека, для больного пища становится настоящим ядом. Без соблюдения диеты приводит к тяжелым последствиям: олигофрении, слабоумию, идиотии.
Механизм развития заболевания

Передается болезнь исключительно наследственным путем и является одной из немногих генетических патологий, которые поддаются лечению. Родиться с этим диагнозом можно только в том случае, если оба родителя носители гена ФКУ.
Связано заболевание с недостаточностью печеночных ферментов, которые участвуют в переработке аминокислоты в тирозин. В результате фениланин накапливается в жидкостях, тканях и оказывает токсическое воздействие на организм.
Симптомы фенилкетонурии
Распознать заболевание у новорожденного ребенка без специальных анализов невозможно. Первые признаки возникают, когда начинается кормление грудью или смесью. При этом симптомы неспецифичные и их сложно идентифицировать. Их с трудом распознают как родители, так и врачи. Сначала появляется необоснованная слабость, беспокойство. Ребенок мало двигается, практические не улыбается, присутствует блуждающий взгляд. К полугоду отстает в умственном, физическом развитии, слабо реагирует на окружающих и происходящее вокруг. К 1-2 годам не выражает свои эмоции мимикой, голосом, не понимает речь и обращение взрослых. У детей постарше выявляют олигофрению, нарушение мышечной спастики.
- необычный запах тела, связанный с аномальным составом пота, мочи;
- светлый цвет волос, кожи, глаз;
- экзематозные изменения эпидермиса;
- согнутые нижние и верхние конечности в суставах.
Методы диагностики

Для всех новорожденных детей в обязательном порядке диагностика этого заболевания проводится в роддоме. Скрининг-тест осуществляют через 72 часа жизни ребенка, если он рожден в срок, и через неделю у недоношенных детей. Проводится забор крови из пятки. Образцы наносят на бланк-фильтр, после чего исследуют концентрацию фениланина. Если показатель превышает 2.2 мг, проводят дополнительные обследования для уточнения диагноза. Также проводят биохимическое исследование мочи (определяют наличие кетоновых тел).
При появлении клинических симптомов фенилкетонурии и для прогноза вероятности развития патологии у будущих детей назначают генетическое исследование мутации гена РАН.
Помимо определения концентрации фениланина также проводят: тест Гатри (предоставляет данные о накоплении аминокислоты в тканях), проба Феллинга (хлорид-железа и уксусную кислоту добавляют в образец мочи, при положительном результате она принимает зеленый, оливковый оттенок).
- биопсия печени для определения активности фермента, ответственного за распад фениланина;
- ЭЭГ, МРТ головного мозга для выявлений отклонений от нормы в структурах мозга.
Лечение
Лечение заключается в строгом контроле питания и ограничении употребления аминокислоты до безопасной нормы. Для новорожденных и грудных детей разработаны специальные смеси, например, Лофенилак. Для более взрослых детей основу рациона должны составлять низкобелковые продукты. Также назначают прием витаминно-минеральных комплексов. При наличии показаний ―ноотропы, антиконвульсанты.
Единый Call-центр 8 800 700 4728
© 2015-2023 ООО “МЕДИЦИНСКАЯ ЛАБОРАТОРИЯ “ОПТИМУМ”. Все права защищены
Россия, Краснодарский край,
г. Сочи, ул. Старонасыпная, 22.
Размещенная на сайте информация и прейскурант не являются публичной офертой.
Источник https://www.medicina.ru/patsientam/zabolevanija/fenilketonuriya/
Источник https://medside.ru/fenilketonuriya
Источник https://analizy-sochi.ru/vrozhdennye-anomalii/fenilketonuriya-fku.html