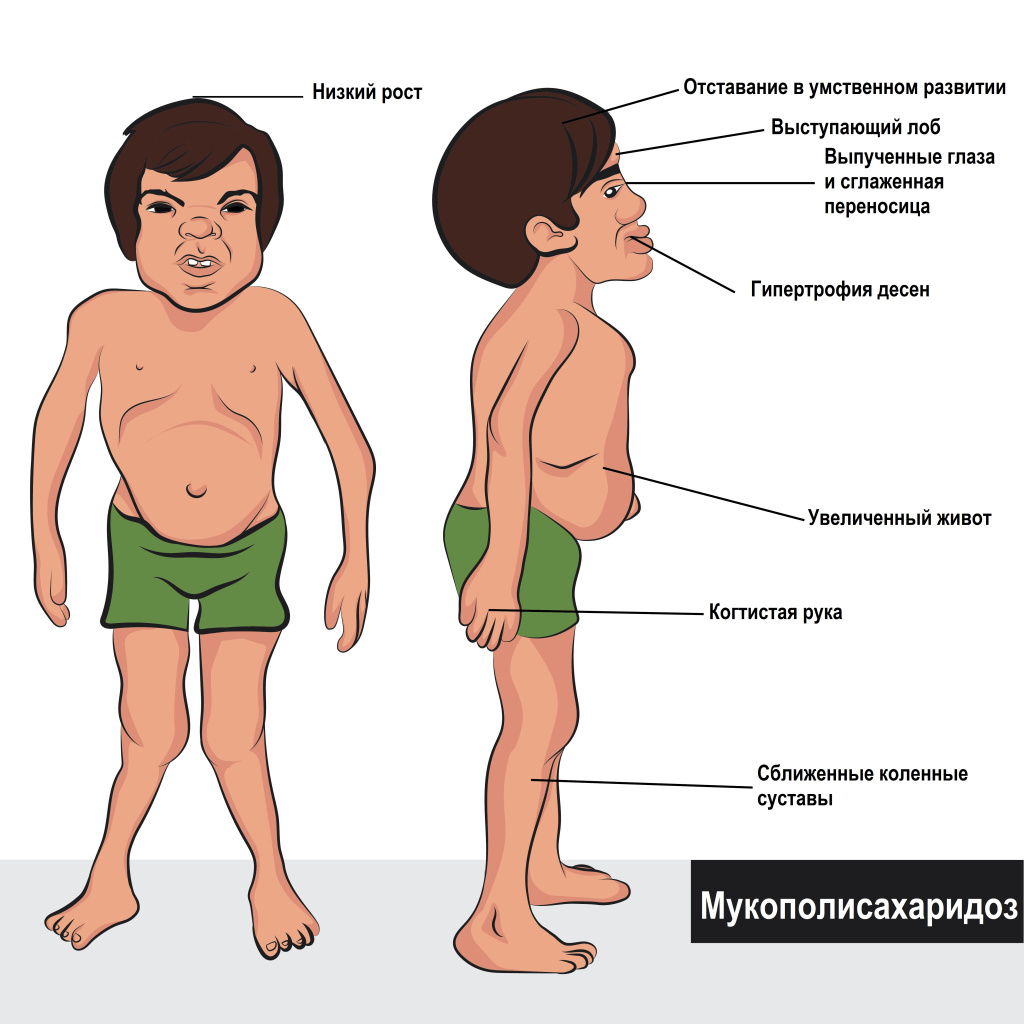
Мукополисахаридоз
Мукополисахаридозы – это группа генетически обусловленных заболеваний, возникающих вследствие нарушения обмена кислых мукополисахаридов (гликозаминогликанов). Характерны системные поражения скелета и задержка физического развития. При некоторых формах наблюдается умственная отсталость. Возможны нарушения сердечной деятельности, патология органов зрения, образование грыж, неврологические нарушения, гипертрихоз, увеличение печени и селезенки. Диагноз выставляется на основании клинических признаков, данных рентгенографии и других исследований. Лечение симптоматическое.
МКБ-10
E76.0 E76.1 E76.2
- Виды мукополисахаридоза
- Мукополисахаридоз типа IH
- Мукополисахаридоз типа I-S
- Мукополисахаридоз типа II
- Мукополисахаридоз типа III
- Мукополисахаридоз типа IV
- Другие типы мукополисахаридоза
Общие сведения
Мукополисахаридозы – группа генетических заболеваний, сопровождающихся накоплением кислых мукополисахаридов в органах и тканях. Причиной развития является передающаяся по наследству неполноценность лизосомных ферментов. Впервые мукополисахаридоз был описан Гурлер в 1917 году. Лечение мукополисахаридоза осуществляют травматологи-ортопеды при участии кардиологов, офтальмологов, неврологов, отоларингологов и других специалистов.
Виды мукополисахаридоза
Мукополисахаридоз типа IH
Мукополисахаридоз типа IH или синдром Гурлер встречается у 1 новорожденного из 20-25 тысяч. Симптомы мукополисахаридоза появляются в течение первого года жизни, полная клиническая картина формируется к 1-2 годам. Для данной формы мукополисахаридоза характерны грубые черты лица и деформация черепа в форме лодочного киля (скароцефалия). Из-за увеличенных аденоидов и пороков развития в области лица и носа больные дышат ртом. Отмечаются прогрессирующие деформации конечностей и других частей скелета, отставание в росте.
Позвоночник пациентов с мукополисахаридозом искривлен, из-за чего в положении сидя возникает симптом «кошачьей спины». Отмечается укорочение шеи, высокое расположение лопаток и выстояние нижних ребер. Кисти широкие, напоминающие когтистую лапу. Со временем у больных мукополисахаридозом формируются контрактуры суставов. Вначале поражаются локтевые и плечевые суставы, затем – коленные, тазобедренные и голеностопные. Из-за ограничения движений возникает характерная походка – на цыпочках с полусогнутыми ногами.
Тоны сердца приглушены, границы расширены. При аускультации определяются систолические шумы, на ЭКГ выявляется диффузное поражение миокарда. Передняя брюшная стенка пациентов с мукополисахаридозом ослаблена, живот увеличен, часто выявляются гидроцеле, пупочные и паховые грыжи. Печень и селезенка увеличены. Характерны нарушения зрения и слуха. Возможно помутнение и увеличение роговицы, пигментная дистрофия сетчатки, глаукома, атрофия зрительных нервов и застойные явления в области глазного дна. У больных мукополисахаридозом этого типа развивается прогрессирующая умственная отсталость. Могут возникать нарушения координации движений, парезы и параличи. Выявляется избыточный рост пушковых волос.
Рентгенография позвоночника пациентов с мукополисахаридозом свидетельствует о характерной деформации позвонков. Позвонки имеют кубовидную форму, их контуры закруглены, в переходном отделе выявляется скошенность передневерхних углов, углообразный кифоз, утолщение и укорочение отростков. На рентгенографии грудной клетки определяется утолщение передних и истончение задних отделов ребер, сопровождающиеся их лопатовидной или саблевидной деформацией, укорочение и деформация ключиц, уменьшение и смещение головок плечевых костей.
Рентгенография таза подтверждает сужение тазового кольца, скошенность вертлужных впадин и уменьшение головок бедренных костей. На рентгенограммах трубчатых костей выявляется истончение кортикального слоя и расширение костномозгового канала. Рентгенография костей кисти свидетельствует о недоразвитии ногтевых фаланг, укорочении и расширении пястных костей, проксимальных и средних фаланг. На рентгенограммах черепа определяется недоразвитие костей лицевого черепа, краниостеноз и макроцефалия.
Мукополисахаридоз типа I-S
Мукополисахаридоз типа I-S или болезнь Шейе (описана в 1962 г. американским офтальмологом Шейе) является более поздним, относительно благоприятно протекающим вариантом мукополисахаридоза типа IH (синдрома Гурлер). До 3-6 лет развитие детей соответствует норме. Первым признаком мукополисахаридоза становятся сгибательные контрактуры пальцев рук. В последующем ограничивается разгибания в лучезапястных, локтевых и плечевых суставах. Контрактуры нижних конечностей, как правило, слабо выражены. Полная клиническая картина мукополисахаридоза формируется к началу подросткового возраста.
Пациенты с мукополисахаридозом коренастые, невысокие, с грубыми чертами лица и хорошо развитой мускулатурой. Отмечается повышенное оволосение (гипертрихоз). Часто возникают паховые или пупочные грыжи. Кожа на пальцах натянута и утолщена. Из-за сдавления срединного нерва возможно развитие синдрома запястного канала, сопровождающееся атрофией мышц области тенара и парестезиями в области III-IV пальцев. У некоторых больных мукополисахаридозом выявляется аортальный стеноз, недостаточность клапанов аорты, пигментная дистрофия сетчатки, глаукома и помутнение роговицы. Интеллект в норме, увеличение селезенки и печени нехарактерно. На рентгенограммах определяется картина, аналогичная мукополисахаридозу типа IH, но патологические изменения выражены менее резко.
Мукополисахаридоз типа II
Мукополисахаридоз типа II или синдром Хантера чаще выявляется у мальчиков и обычно развивается на 2-3 году жизни. Как и при мукополисахаридозе типа IH наблюдается скафоцефалия, огрубление черт лица, низкий голос и затруднения дыхания из-за деформации лицевого скелета. При этом характерный для мукополисахаридоза типа IH кифоз обычно не выявляется, симптом «кошачьей спины» отрицательный. Пациенты часто страдают от респираторных инфекций (бронхитов, трахеитов, пневмоний). Постепенно развиваются нарушения координации движений, возрастает агрессивность. Настроение неустойчивое, с резкими перепадами.
Также при данной форме мукополисахаридоза наблюдается незначительное увеличение селезенки и печени, прогрессирующая тугоухость, узелки на коже спины и некоторое снижение интеллекта. В последующем может развиться помутнение роговицы. Рентгенологическая картина – как при мукополисахаридозе типа I-S. Возможны два варианта развития болезни: благоприятный (вариант В) и неблагоприятный (вариант А). При благоприятном варианте симптомы выражены нерезко, иногда возникает незначительная умственная отсталость, пациенты с мукополисахаридозом могут доживать до 30 и более лет. Для неблагоприятного варианта характерна яркая клиническая картина и серьезные нарушения интеллекта. Летальный исход наступает в подростковом возрасте.
Мукополисахаридоз типа III
Мукополисахаридоз типа III или болезнь Санфилиппо (описана в 1963 г. американским педиатром Санфилиппо) развивается у 1 новорожденного из 100-200 тысяч. В первые годы жизни развитие соответствует возрасту, иногда наблюдаются затруднения глотания и неуклюжая походка. В возрасте 3-5 лет ребенок с мукополисахаридозом становится апатичным и начинает отставать в развитии. Возникают нарушения речи, огрубление черт лица, недержание мочи и кала. Со временем умственная отсталость прогрессирует и занимает центральное положение в клинической картине этого типа мукополисахаридоза.
Наряду с нарушениями интеллекта наблюдается умеренное увеличение печени и селезенки, повышенное оволосение, контрактуры и задержка роста. Патология со стороны глаз и сердечно-сосудистой системы для этой формы мукополисахаридоза нехарактерна. Рентгенологическая картина – без изменений или как при мукополисахаридозе типа I-S, но менее выраженная. Больные мукополисахаридозом третьего типа, как правило, погибают в возрасте 10-20 лет от инфекционных осложнений.
Мукополисахаридоз типа IV
Мукополисахаридоз типа IV или болезнь Моркио (описана в 1929 г. уругвайским педиатром Моркио) наблюдается у 1 новорожденного из 40 тысяч. До 1-3 лет дети развиваются нормально. В последующем возникает значительное отставание в росте, укорочение шеи и туловища, контрактуры и вальгусная деформация конечностей, сколиоз или кифоз, разнообразные деформации грудной клетки, снижение силы мышц, утолщение кожи и огрубление черт лица. При этом типе мукополисахаридоза часто развиваются паховые и пупочные грыжи, дистрофия роговицы и тугоухость. Интеллект сохранен.
На рентгенограммах позвоночника определяется кифоз, сколиоз, расширение и уплощение тел позвонков. При проведении рентгенографии таза и конечностей выявляются множественные деформации, неровность контуров, уплощение головок бедренных костей, укорочение костей предплечья и деформации стоп. Средняя продолжительность жизни больных мукополисахаридозом – менее 20 лет. Смерть при данной форме мукополисахаридоза наступает из-за сопутствующих заболеваний, осложняющихся сердечно-легочной недостаточностью.
Другие типы мукополисахаридоза
Мукополисахаридоз типа VI или болезнь Марото-Лами (описана в 1960 г. французами Лами и Марото) развивается в возрасте 2 года и старше. Возникает огрубление черт лица, отставание в росте, укорочение шеи, контрактуры суставов и бочкообразная деформация грудной клетки. Характерны частые простуды. Возможны грыжи, увеличение печени и селезенки. Интеллект не страдает. Наблюдается два варианта течения: классический и мукополисахаридоз со слабой выраженностью клинических проявлений. На рентгенограммах больных мукополисахаридозом определяется кубовидная или клиновидная деформация позвонков, треугольная деформация таза, недоразвитие и деформация головок бедренных костей, укорочение малоберцовых костей.
Мукополисахаридоз типа VII протекает, как мупоколисахаридоз типа III, различия выявляются только при проведении биохимических исследований. Мукополисахаридоз типа VIII по симптомам напоминает мукополисахаридоз типа IV, но, в отличие от него, сопровождается задержкой умственного развития.
Диагностика
Диагноз мукополисахаридоза устанавливается на основании характерной клинической и рентгенологической картины, выявления гликозаминогликанов в моче, изучения активности ферментов в клеточных культурах, генетического секвенирования. В ходе обследования больным мукополисахаридозом назначаются консультации различных специалистов: кардиолога, гастроэнтеролога, офтальмолога, отоларинголога, невролога, психиатра и т. д., проводятся инструментальные исследования для оценки состояния различных органов и систем.
Лечение мукополисахаридоза
Патогенетическая терапия не разработана. Лечение симптоматическое, может быть как консервативным, так и оперативным. Осуществляется профилактика и лечение респираторных инфекций, проводится коррекция нарушений зрения и слуха. При необходимости выполняются грыжесечение и герниопластика, операции по устранению контрактур и коррекции деформаций скелета.
Прогноз и профилактика
Прогноз при всех типах мукополисахаридоза неблагоприятный – продолжающееся накопление продуктов обмена в тканях приводит к усугублению патологических изменений со стороны всех органов и систем. Использование любых лечебных средств (переливания крови, введение гормонов и т. д.) при мукополисахаридозе обеспечивает лишь временное улучшение. Рекомендуется пренатальная профилактика.
Литература
1. Руководство по педиатрии. Врожденные и наследственные заболевания/ Новиков П.В., Баранов А.А., Каганов Б.С., Шиляев Р.Р — 2007
Мукополисахаридоз — симптомы и лечение
Что такое мукополисахаридоз? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой Ольги Игоревны, генетика со стажем в 8 лет.
Над статьей доктора Боровиковой Ольги Игоревны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина

Генетик Cтаж — 8 лет
«Evi Clinic» («Эви Клиник»)
Клиника «OXY-center»
Медицинский центр «Андромед»
Дата публикации 24 декабря 2019 Обновлено 26 апреля 2021Определение болезни. Причины заболевания
Мукополисахаридозы — это группа редких наследственных заболеваний соединительной ткани, связанных с нарушением обмена веществ. Они обусловлены нехваткой определённых ферментов, которые участвуют в переработке (расщеплении) гликозаминогликанов — сложных молекул сахара. В связи с чем эти молекулы скапливаются в организме человека в опасно большом количестве и приводят к различным изменениям [1] [14] .
Самые явные проявления мукополисахаридозов — множественные деформации костей и суставов и нарушение физического развития (задержка и утеря ранее приобретённых навыков. При определённых типах заболевания (синдромах Шейе, Гурлер — Шейе, Хантера, Санфилиппо — I, II, III типах мукополисахаридозов) помимо прочего нарушается умственное развитие, начиная с лёгких когнитивных нарушений и заканчивая глубокой деменцией [13] .
Изменения при мукополисахаридозах возникают в результате дефекта ферментного расщепления углеводной части молекулы мукополисахаридов (гликозаминогликанов). При этом в фибробластах и мезенхимальных клетках, которые способны трансформироваться в хрящевые, костные либо жировые клетки, накапливается хондроитинсульфат — вещество, являющееся основой хрящей. Это ведёт к нарушению структуры соединительной, костной и хрящевой ткани [13] .
Мукополисахаридозы встречаются очень редко: примерно у одного ребёнка среди 56-325 тысяч новорождённых. Их причиной является мутация . Эти болезни довольно сложно диагностировать из-за малой осведомлённости врачей о них. Поэтому больным часто выставляют другие диагнозы и проводят неадекватное лечение.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы мукополисахаридоза
Мукополисахаридозы делятся на несколько типов. Они различаются первичным генным дефектом, патологическим ферментом, преимущественным поражением той или иной системы органов и тканей, возрастом начала заболевания и тяжестью его течения (см. классификацию ). В целом при этих генетических патологиях встречаются множественные нарушения: поражаются костная система, хрящи, печень, селезёнка, головной мозг, роговица глаза, органы лимфатической и дыхательной системы. Из-за особенностей строения дыхательных путей возникают частые инфекционные заболевания органов дыхания и слуха, что приводит к развитию тугоухости и респираторным расстройствам — бронхитам, пневмониям и др.
К самым частым проявлениям мукополисахаридозов относят низкорослость, волосатость и нарушения развития. Они дают о себе знать с раннего детства. Лицо ребёнка, как правило, приобретает грубые черты, становится «взрослым», а голова выглядит довольно большой из-за широкого лба и короткого носа. Губы и язык тоже становятся больше, чем у сверстников. Суставы пальцев плохо гнутся и двигаются. При отдельных типах заболевания нарушается слух и зрение, повреждаются сердечные клапаны и артерии [14] .
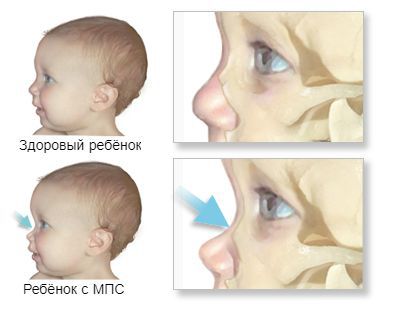
Патогенез мукополисахаридоза
Все формы мукополисахаридоза наследуются по аутосомно-рецессивному типу, т. е. мутировавший ген должен быть у обоих родителей. Исключением является мукополисахаридоз III типа: он наследуется по Х-сцепленному рецессивному типу.
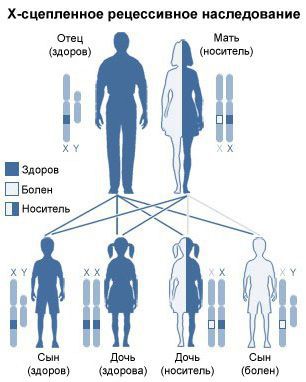
Каждая форма мукополисахаридоза отличается нарушением выработки различных ферментов, что приводит к изменению функции фермента, его нехватке или отсутствию. В результате этого нарушается ферментозависимый распад гликозаминогликанов: из-за нехватки определённых ферментов организм не в состоянии преобразовывать и сохранять эти полисахариды в тканях. Нерасщеплённые гликозаминогликаны через кровь распространяются по всему организму, что приводит к их избыточному накоплению в различных органах и системах [14] . Чаще всего эти вещества скапливаются в соединительной ткани, сердце, печени, селезёнке, нервной ткани и оболочках мозга:
- Отложение гликозаминогликанов в оболочках мозга способствует сужению субарахноидальных пространств, которые находятся под паутинной оболочкой головного мозга, и формированию гидроцефалии .
- Поражение нервных клеток приводит к задержке умственного развития, прогрессирующей деменции.
- Деформация метафизов (отделов трубчатых костей), утолщение межсуставных хрящей и суставных связок становятся причинами нарушения подвижности в суставах.
- В результате отложения гликозаминогликанов в миндалинах, трахее и голосовых связках возникает отёчность верхних дыхательных путей, нарушается дыхание, что приводит к нарушению вентиляции, частым респираторным заболеваниям, отитам, обструктивным состояниям (непроходимости дыхательных путей).
- Отложение гликозаминогликанов в тканях сердца сопровождается кардиомиопатиями и формированием клапанных пороков сердца.
- Повреждение канальцев почек может приводить к прогрессирующей артериальной гипертензии.
Классификация и стадии развития мукополисахаридоза
В зависимости от первичного генетического дефекта выделяют несколько типов мукополисахаридоза:
- МПС I типа включает в себя несколько подтипов: синдром Гурлер (H), Шейе (S) и Гурлер — Шейе (H/S) [6][7] . Мутация при данных фенотипах обнаруживается в гене IDUA, вызывает дефицит фермента альфа-L-идуронидазы [4] .
- МПС II типа — синдром Хантера [8] . Мутация при данном заболевании обнаруживается в гене IDS, вызывает дефицит либо отсутствие идуронат-2-сульфатазы, либо дефицит или отсутствие сульфоидуронат сульфатазы [5] .
- МПС III типа — синдром Санфилиппо — и меет несколько подтипов:
- МПС III А обусловлен мутацией в гене SGSH, приводит к нехватке гепаран-N-сульфатазы [9] ;
- МПС III В обусловлен мутацией в гене NAGLU, вызывает дефицит N-ацетил-α-D-глюкозаминидазы [10] ;
- МПС III С обусловлен мутацией в гене HGSNAT, вызывает дефицит гепаран-α-глюкозаминид-N-ацетилтрансферазы [11] ;
- МПС III D обусловлен мутацией в гене GNS, вызывает дефицит N-ацетилглюкозамин-6-сульфатазы [2][12] .
- МПС IV типа — синдром Моркио — делится на два подтипа:
- МПС IV А связан с мутацией в гене GALNS, вызывает дефицит галактозамин-6-сульфатазы;
- МПС IV В связан с мутацией в гене GLB1, вызывает недостаточность β-галактозидазы.
- МПС VI типа — синдром Марото — Лами — связан с мутацией гена ARSB, вызывает дефицит N-ацетилгалактозамин-4-сульфатазы [3] .
- МПС VII типа — синдром Слая — возникает вследствие мутации гена GUSB, приводит к дефициту β-глюкуронидазы.
- МПС IX типа является проявлением мутации гена HYAL1, приводит к недостаточности гиалуронидазы.
Остановимся подробнее на каждом типе мукополисахаридоза.
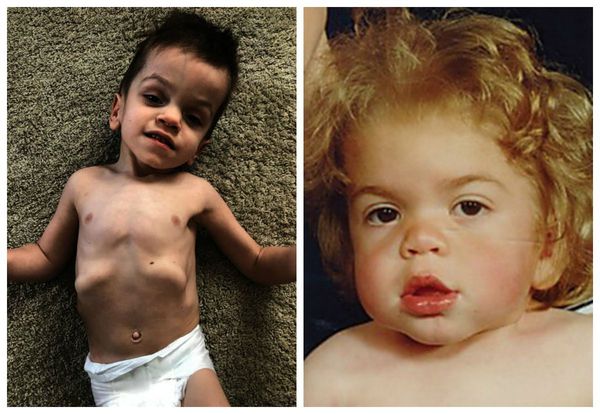
Мукополисахаридоз I H (синдром Гурлер). Основными симптомами заболевания являются: задержка психического и физического развития, умственная отсталость, грубые черты лица, пороки клапанов сердца, помутнение роговицы, низкорослость, тугоподвижность суставов. Первые признаки такого мукополисахаридоза появляются в течение первых 12 месяцев жизни младенца.
Иногда уже с самого рождения отмечается увеличение печени и селезёнки (гепатомегалия), пупочные или пахово-мошоночные грыжи. Ближе к 6-12 месяцам лицо приобретает грубые черты, напоминающие гаргулью: голова становится больше, выступают лобные бугры, появляются широкие скулы, уплощается и втягивается переносица, укорачиваются носовые ходы, изменяется форма носа, ноздри выворачиваются, рот постоянно полуоткрыт, увеличивается язык, губы становятся пухлыми. Низкорослость становится заметной ближе к 2-5 годам, рост обычно ниже 100 см. Пропорции тела нарушены, шея укорочена [3] .
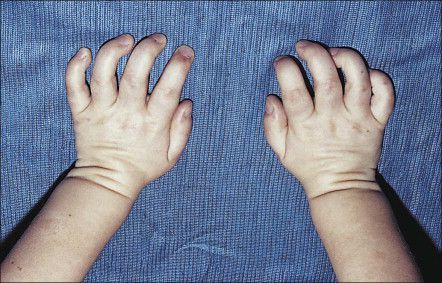
Очень часто у таких пациентов уменьшается подвижность крупных и мелких суставов, особенно пальцев, укорачиваются трубчатые кости, кисти рук деформируются и приобретают форму «лапы с когтями» [1] . Уменьшаются головки бедренных костей, из-за чего формируется дисплазия тазобедренных суставов. Подвздошные кости приобретают треугольную форму.
С развитием болезни к клинической картине присоединяются признаки поражения внутренних органов, сердца и сосудов, головного мозга, нервной системы.
Со стороны сердечно-сосудистой системы утолщаются клапаны, уменьшается диаметр артерий, нарушается сократимость сердечной мышцы, возникают кардиомиопатии, повышается артериальное давление. В итоге развивается сердечная недостаточность .
Основными проявлениями вовлечения нервной системы являются прогрессирующее снижение умственных способностей, задержка лингвистического (языкового) развития, гипотония мышц, снижение сухожильных рефлексов, патология черепно-мозговых нервов, снижение слуха [6] . Психическое и моторное развитие запаздывает, достигает максимального развития на уровне 2-4 лет, после чего останавливается и регрессирует, иногда переходя в полную деменцию. Ухудшает ситуацию прогрессирующая гидроцефалия [1] .
Поведение пациентов с синдромом Гурлер страдает из-за когнитивных нарушений, прогрессирующей тугоухости, бессонницы, связанной с ночной обструктивной задержкой дыхания . Со временем ребёнок становится всё активнее, развивается синдром дефицита внимания , агрессия, расстройства аутистического спектра.
При деформации позвоночника, утолщении оболочек спинного мозга изменяется походка, возникает мышечная гипотония, нарушается чувство равновесия, отмечается непроизвольное мочеиспускание и задержка мочи. При тяжёлом течении заболевания возможны судороги, требующие применения антиконвульсантов.
В возрасте 5-10 лет и старше часто развивается синдром запястного канала . Данное состояние требует обязательной коррекции, в противном случае развиваются контрактуры дистальных межфаланговых суставов, пальцы теряют чувствительность, наступает парез мышц большого пальца.
Скопление полисахаридов в глоточном лимфоидном кольце, надгортаннике и трахее является причиной сужения дыхательных путей, развития обструктивных состояний, рецидивирующих инфекций дыхательных путей и среднего уха.
Для больных мукополисахаридозом I типа также характерно поражение глаз в виде прогрессирующего помутнения роговицы, пигментной дегенерации и повышения внутриглазного давления [1] .
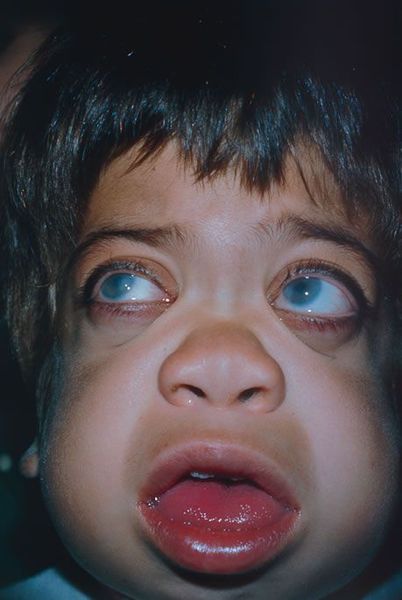
Средняя продолжительность жизни детей с синдромом Гурлер достигает 10 лет. Частой причиной смерти становятся обструктивные заболевания дыхательной системы, острые и хронические инфекционные процессы, патология сердечно-сосудистой системы.
Мукополисахаридоз I H/S (синдром Гурлер — Шейе) является среднетяжёлой формой заболевания. К самым частым симптомам относятся нарушение движения в суставах, снижение прозрачности роговицы и низкий рост [7] .
Первые признаки проявляются к 3-8 годам. Из-за преждевременного сращения черепных швов изменяется форма черепа и нарушается рост головного мозга. Переносица уплощается и западает, губы становятся пухлыми, верхняя челюсть становится меньше, повышается оволосение, кожа утолщается.
Как правило, в течение первых 12 месяцев жизни рост нормальный, но по мере взросления скорости роста снижается и развивается низкорослость. Пропорции тела нарушаются [1] . Суставы становятся тугоподвижными, нарушается форма костей, деформируется грудная клетка, возникает кифоз, сколиоз позвоночника.
С возрастом увеличиваются миндалины, рецидивируют респираторные заболевания, воспаления среднего уха, нарушается проходимость дыхательных путей. Со стороны глаз поражается роговица: происходит её помутнение из-за накопления гликозаминогликанов [7] . Нарушение мозга сопровождается задержкой психического, моторного, лингвистического развития, в исходе развивается деменция. Отмечается гидроцефалия, пахименингит в верхних отделах позвоночника, что приводит к компрессии спинного мозга, развитию патологии в нижележащих отделах [1] . Развивается синдром запястного канала, являющийся причиной контрактур суставов. Со стороны сердечно-сосудистой системы развиваются клапанные аномалии за счёт утолщения створок, нарушение сократимости сердечной мышцы. Увеличение печени и селезенки приводит к диспропорциональному увеличению живота. Патология передней брюшной стенки проявляется пахово-мошоночными грыжами, широким пупочным кольцом с образованием грыж [6] .
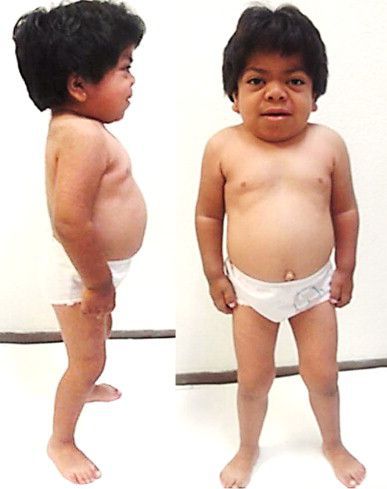
Мукополисахаридоз I S (синдром Шейе, или болезнь Гурлер с поздним началом) — лёгкая форма заболевания. Отличается стёртой клинической картиной. Грубые черты лица, напоминающие гаргулью, также присутствуют, но в более лёгкой степени, чем при синдроме Гурлер. Интеллект, психическое и моторное развитие, как правило, в пределах нормы. Иногда наблюдается незначительное запаздывание развития.
Основными клиническими проявлениями являются поражения суставов, сопровождающиеся нарушением их функции, и отставание в росте [1] . Поражение костей и суставов приводит к тугоподвижности конечностей, болевому синдрому, деформации кистей по типу «лапы с когтями», «пустой стопы», вальгусу в коленных суставах (Х-образным голеням), тоннельный синдром способствует развитию контрактур.
Характерны увеличение печени, селезёнки, образование пахово-мошоночных грыж, слабости пупочного кольца [1] , болезни органов слуха и рецидивирующие инфекционные заболевания органов дыхательной системы, которые приводят к приступам ночного апноэ. Зрение страдает из-за неравномерного помутнения роговицы, повышения внутриглазного давления, пигментной дегенерации сетчатки, что более характерно для больных старше 30 лет.
Часто формируется компрессия срединного нерва, что приводит к развитию карпального туннельного синдрома, связанного со сдавлением срединного нерва между костями, мышцами и связками запястья. Клапанные пороки сердца, расширения аорты могут быть причиной развития сердечной недостаточности.
Мукополисахаридоз II типа (синдром Хантера) бывает тяжёлой и умеренно тяжёлой формы. Такое разделение основано на степени поражения нервной системы и возрасте, в котором проявляются первые признаки болезни.
Синдром Хантера включает в себя множество различных признаков с поражением различных органов и систем [8] . Основу клинической картины составляют нарушения центральной нервной системы, проявляющиеся задержкой умственного развития, огрубление черт лица, низкорослость, нарушение подвижности суставов.
Большинство симптомов схожи с I типом болезни: грубые черты лица, напоминающие гаргулью, увеличенная мозговая часть черепа, выступающий лоб, уплощённая запавшая переносица, плоский нос с укороченными носовыми ходами, выворачивающиеся ноздри, приоткрытый рот, увеличенный язык, толстые губы, низкий рост, укороченная шея, тугоподвижность суставов, повышенное оволосение, пахово-мошоночные и пупочные грыжи. Отличается запоздалым прорезыванием зубов, длинными густыми ресницами, широкими густыми сросшимися бровями [8] . При прогрессировании заболевания волосы осветляются и выпрямляются, становятся сухими и жёсткими.
Для синдрома Хантера характерна сыпь в виде мелких узелков, группирующихся на спине, плечах и бёдрах. Её появление связано со скоплением мукополисахаридов в дерме.
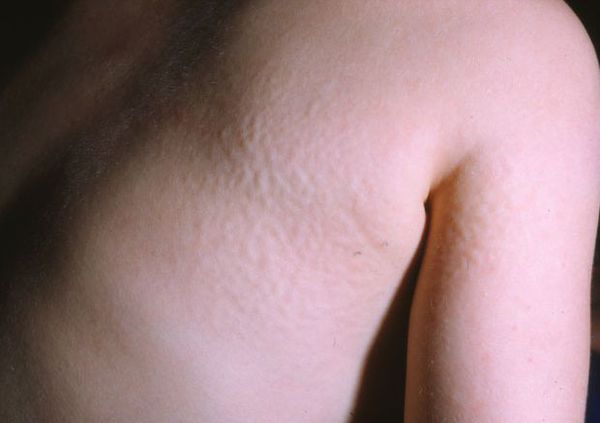
За счёт укорочения и расширения пястных костей, развития пястного тонельного синдрома кисть приобретает когтистую форму. По мере прогрессирования заболевания формируется кифосколиоз, деформируются тазобедренные суставы, возникают различные костные аномалии, увеличивается размер турецкого седла — углубления в клиновидной кости черепа.
Рецидивирующие инфекции органов дыхания и слуха, увеличение миндалин приводят к тугоухости, апноэ во сне. Поражение глаз проявляется помутнением роговицы (реже, чем при других типах болезни), пигментной дегенерацией сетчатки, а при тяжёлых формах происходит дистрофия сетчатки и отёк диска. При развитии внутричерепной гипертензии наблюдается отёк зрительного нерва.
Психическое развитие начинает отставать в возрасте 1,5-3 лет. При тяжёлых формах заболевания к 8-10 годам появляется тяжёлая умственная отсталость и эпилепсия, которая трудно поддаётся лечению. Иногда формируется сообщающаяся гидроцефалия, парезы, потеря чувствительности в конечностях. Известны случаи сдавления спинного мозга, связанные с увеличением толщины оболочек, нестабильностью атлантоаксиального сустава (между затылочной костью и первым шейным позвонком). Это приводит к мышечной слабости, недержанию мочи, задержке мочеиспускания и неуклюжести [6] . Из-за поражения ствола головного мозга нарушается глотание и подвижность нижней челюсти. Возможны псевдобульбарные и бульбарные параличи (связанные с поражением продолговатого мозга).
Большую трудность представляют поведенческие нарушения: гиперактивность, агрессия, упрямство. Часто ухудшают ситуацию проблемы со сном, нарушения слуха. Со временем присоединяются расстройства аутистического спектра. Эти состояния трудно поддаются коррекции. При тяжёлом течении заболевания развивается деменция.
Поражение сердечно-сосудистой системы проявляется клапанными пороками сердца, кардиомиопатией.
С раннего детского возраста отмечается увеличение печени и селезёнки, нарушение переваривания пищи и моторики кишечника. Увеличение языка и поражение височно-нижнечелюстного сустава приводят к нарушению глотания.
Мукополисахаридоз III типа (синдром Санфилиппо) отличается от других тем, что при нём гликозаминогликаны скапливаются по большей части в тканях мозга, а не в соединительной ткани. Самые яркие клинические признаки: задержка психического развития, маловыраженное поражение суставов, лёгкое огрубление черт лица [2] .
Первые проявления возникают на втором году жизни: низкорослость, умеренная тугоподвижность суставов, увеличение печени и селезёнки [9] . До 3 лет дети обычно развиваются в пределах нормы, а затем утрачиваются ранее приобретённые моторные и речевые навыки, нарушается психическое развитие. В дальнейшем возникает нарушение поведения, грубое нарушение психики, переходящее в деменцию. Речь, как правило, не формируется.
Характерны черепно-лицевые изменения: увеличенные лобные бугры, низкие надбровные дуги, широкая спинка носа, густые сросшиеся брови, густые ресницы, сухие и жёсткие волосы, повышенное оволосение [2] .
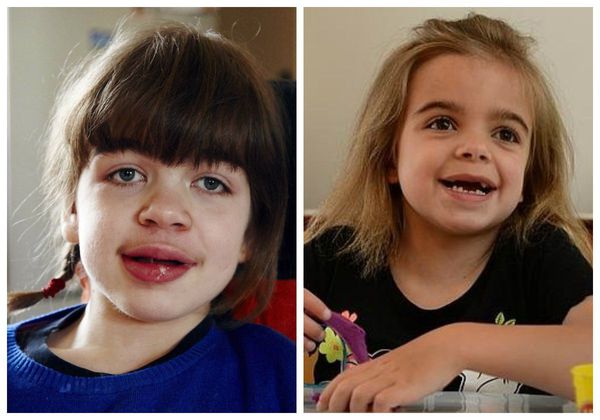
Мукополисахаридоз IV типа (синдром Моркио) сопровождается значительными деформациями костной системы, которые затрагивают в основном руки, ноги и грудную клетку.
В первые месяцы жизни ребёнка признаки заболевания отсутствуют, клиническая картина проявляется только в возрасте 1-3 лет. К 7-8 годам симптомы болезни становятся наиболее яркими. Характерна низкорослость, задержка физического развития. Кожа толстая, малоэластичная. Лицевые признаки: широкий рот, укороченный нос, редкие зубы, дисплазия эмали зубов.
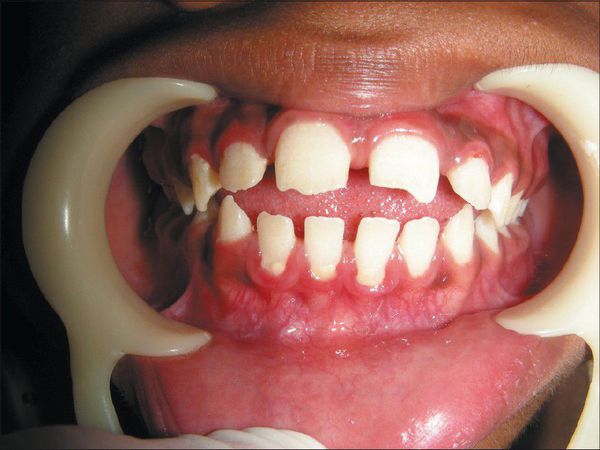
Мышцы гипотоничны, грудная клетка деформирована, отмечается кифосколиоз грудного и поясничного отделов позвоночника. Интеллект не страдает [3] . Суставы тугоподвижны, нередко развиваются контрактуры. Часто появляется шейная миелопатия — поражение волокон спинного мозга с нарушением проведения нервного импульса. Иногда развивается тугоухость. Отмечается слабость апоневроза передней брюшной стенки с образованием грыж [3] .
Мукополисахаридоз VI типа (синдром Марото — Лами) имеет разнообразные проявления, которые прогрессируют с разной скоростью. Характерна низкорослость, снижение зрения, грубые черты лица, тугоухость, снижение подвижности суставов, увеличение печени и селезёнки, поражение сердечно-сосудистой системы и органов дыхания. Интеллект, как правило, в пределах нормы.
Черты лица напоминают гаргулью. Нарушены пропорции тела, формируется карликовость. Суставы деформированы, множественные деформации костей приводят к инвалидизации. Отмечается нарушение развития тел грудных позвонков, их переломы при незначительной нагрузке. Часто возникает сдавление спинного мозга, связанное с нестабильностью шейного отдела позвоночника. Иногда развиваются клапанные пороки сердца, приводящие с сердечной недостаточности. Со стороны желудочно-кишечной системы наблюдается синдром раздражённого кишечника, увеличение печени и селезёнки.
Мукополисахаридоз VII типа (синдром Слая) проявляется увеличением печени и селезёнки, образованием пахово-мошоночных или пупочных грыж, низкорослостью, деформацией грудной клетки, кифосколиозом в поясничном и крестцовом отделах позвоночника, искривлением нижних конечностей, рецидивирующими респираторными заболеваниями, грубыми чертами лица с широко расставленными глазами, уплощённой переносицей и вывернутыми вперёд ноздрями. Иногда наблюдаются клапанные пороки сердца и кардиомиопатия.
Осложнения мукополисахаридоза
Основными осложнениями течения мукополисахаридозов различных типов являются тяжёлые рецидивирующие респираторные инфекции (риниты, синуситы, отиты, ОРЗ), приводящие к дыхательной недостаточности, патология сердечно-сосудистой системы и поражение головного мозга.
Сам мукополисахаридоз не приводит к смерти. Больные чаще всего умирают в результате сердечной и дыхательной недостаточности, которые развиваются на фоне заболевания [5] .
Диагностика мукополисахаридоза
Диагностика основана на определении характерных клинических (внешних) признаков и изучении активности гликозаминогликанов в крови. Последнее исследование проводится следующим образом: на специальные бланки с фильтровальной бумагой капается капиллярная кровь пациента, затем высушивается и отправляется в лабораторию, где проводится ферментный анализ. Возможно определение концентрации гликозаминогликанов в моче, но часто встречаются ложные результаты [4] [14] .
Также в рамках диагностики проводится генетическое исследование. Оно заключается в поиске мутаций в определённых генах, отвечающих за развитие мукополисахаридозов.
Дополнительная диагностика заключается в поиске поражений органов и систем с помощью различных исследований:
- УЗИ — выявляет увеличение печени и селезёнки, пороки развития сердца и увеличение его размеров;
- рентгенография — выявляет патологию костей и суставов;
- электрокардиография — выявляет кардиомиопатию, патологию сократимости и проводимости;
- электромиография — позволяет диагностировать нарушения в проведении возбуждения по нервным волокнам к мышцам;
- аудиометрия — помогает выявить проблемы со слухом [5][14] .
При проведении рентгенологических исследований в случае мукополисахаридоза I H типа выставляется диагноз «множественный дизостоз» — нарушения развития костей. Диафизы длинных трубчатых костей расширены, при рентгенологических исследованиях отмечается изменение структуры метафизов и эпифизов. Ключицы короткие и толстые, рёбра приобретают форму весла, части, расположенные ближе к позвоночнику, сужены, а передние — толстые и широкие [1] . Фаланговые кости рук и ног укорочены, имеют форму трапеции, диафизы широкие.
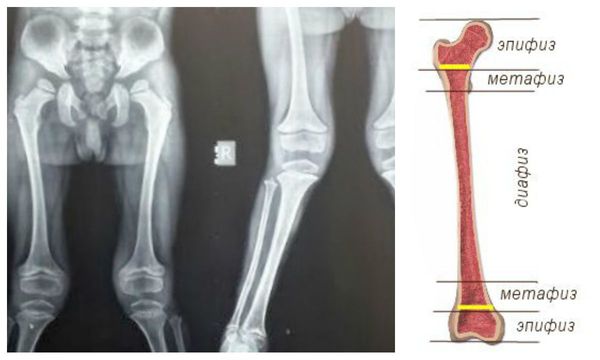
Формируются уплощение позвонков, сколиоз, кифосколиоз. Позвонки в поперечнике широкие, но они низкие. В деформированных участках отмечается недоразвитие поперечных отростков либо их «языкообразная» трансформация.
До рождения мукополисахаридоз и другие хромосомные нарушения можно обнаружить с помощью амниоцентеза (исследования околоплодной жидкости) или биопсии хориона (слоя плаценты). Также риск появления мукополисахаридоза у младенца можно определить ещё до зачатия. Для этого родителям нужно пройти генетический анализ [14] .
Лечение мукополисахаридоза
Симптоматическое лечение заключается в устранении грыжи, удалении миндалин и ортопедической хирургии. Проводится коррекция искривлений позвоночного столба при помощи транспедикулярной фиксации металлическими конструкциями, исправление деформаций костей и суставов, коррекция грудной клетки. Это позволяет облегчить болевой синдром, уменьшить риск сдавления спинного мозга. Также проводится шунтирование желудочков головного мозга при гидроцефалии.
Патогенетическое лечение, направленное на причину болезни, возможно при помощи трансплантации костного мозга и стволовых гемопоэтических клеток и заместительной ферментной терапии [5] . Трансплантация возможна от родственного донора либо из стволовых клеток пуповинной крови родственного донора после проведения химиотерапевтической или лучевой супрессии. Данный вид лечения не получил широкого распространения из-за высокого риска осложнений (инвалидизации или смерти). Заместительная ферментная терапия способна остановить прогрессирование заболевания, частично улучшить уже сформированные патологии. Применение такой терапии позволяет добиться регресса заболевания и существенно улучшить прогноз жизни.
В настоящее время ферментная терапия разработана только для некоторых типов мукополисахаридозов. При I типе используется альдуразим . Он представляет собой рекомбинантную человеческую альфа-L-идуронидазу. Препарат вводится внутривенно каждую неделю в течение 4-х часов [4] . Для мукополисахаридоха II типа показана элапраза [2] .
Мукополисахаридозы относятся к редким заболеваниям, поэтому препараты для их лечения выделяются на государственном уровне и предоставляются пациентам в рамках оказания бесплатной медицинской помощи [5] . В некоторых случаях заместительная терапия является пожизненной [14] .
Прогноз. Профилактика
При отсутствии должного лечения прогноз неблагоприятный, так как больные погибают от осложнений. Практически всегда заболевание приводит к инвалидизации. Однако всё зависит от типа заболевания. В некоторых случаях пациенты могут жить так же долго, как и здоровые люди [14] .
Профилактика заключается в генетическом консультировании пар, вступающих в брак, особенно при отягощённом семейном анамнезе.
Возможно проведение преимплантационной генетической диагностики в циклах ЭКО у пар с высоким риском рождения ребёнка с мукополисахаридозом либо генетическая диагностика на ранних сроках беременности с возможностью прерывания беременности при выявлении заболевания у плода.
Перспективным является создание генетических паспортов, выбор партнёра с отсутствием соответствующих мутаций.
Список литературы
- Министерство здравоохранения РФ. Федеральные клинические рекомендации по диагностике и лечению мукополисахаридоза I типа. — 2013.
- Союз педиатров России. Клинические рекомендации по ведению детей с мукополиса-харидозом III типа. — 2013.
- Союз педиатров России. Федеральные клинические рекомендации по оказанию мади-цинской помощи детям с мукополисахаридозом IV типа. — 2013.
- Министерство здравоохранения РФ. Федеральные клинические рекомендации по ока-занию медицинской помощи детям с мукополисахаридозом I типа. — 2015.
- Министерство здравоохранения РФ. Федеральные клинические рекомендации по диагностике и лечению мукополисахаридоза II типа. — 2013.
- Hamosh A. Hurler syndrome. Mucopolysaccharidosis type IH; MPS1-H // Online Mendelian Inheritance in Man. — 2018
- Przylepa K. A. Scheie syndrome. Mucopolysaccharidosis type IS; MPS1-S. Mu-copolysaccharidosis type V, Formerly, MPS V, Formerly; MPS5, Formerly // Online Mendelian Inheritance in Man. — 2002
- Hamosh A. Mucopolysaccharidosis, type II; MPS2 // Online Mendelian Inher-itance in Man. — 2018.
- Hamosh A. Mucopolysaccharidosis, type IIIA; MPS3A // Online Mendelian In-heritance in Man. — 2018.
- Hamosh A. Mucopolysaccharidosis, type IIIB; MPS3B // Online Mendelian In-heritance in Man. — 2012.
- Kniffin C. L. Mucopolysaccharidosis, type IIIC; MPS3C // Online Mendelian Inheritance in Man. — 2011.
- O’Neill M. J. F. Mucopolysaccharidosis, type IIID; MPS3D // Online Mendelian Inheritance in Man. — 2010.
- Боровикова О. И. Типы мукополисахаридозов и клинические проявления // Счастливая беременность. — 2019.
- Demczko M. Mucopolysaccharidoses // MSD Manuals. — 2018.
Мукополисахаридоз 1, 2 типа: факторы риска развития, характерные симптомы, особенности лечения и прогноз
В этом году исполняется ровно 100 лет с тех пор, как канадец Чарльз Хантер составил первое описание мукополисахаридоза, выявленного у двух братьев. Сегодня известно, что это не одно, а целая группа заболеваний, очень редких.
В мире проживает примерно 1,5 тысячи таких пациентов, около 200 из них живут в России. Мы выясняли, что такое мукополисахаридоз и какова судьбы людей, больных этим заболеванием.

Мукополисахаридоз: ферменты и болезнь
Немалая часть нашего организма состоит из соединительной ткани. Ее клетки участвуют в формировании каркаса и наружного покрова всех органов тела. В некоторых их них соединительная ткань составляет до 90% от их массы. Это хрящи, кости, жир, фасции, синовиальная жидкость, которая плещется в суставах, лимфа, склера и радужка глаза, микроглия, окружающая нейроны и др.
Без преувеличения, главным элементом соединительной ткани можно считать протеогликаны. Это соединения, которые содержат белковое ядро и связанные с ним многочисленные и разнообразные гликозаминогликаны (ГАГ). В разных тканях — разные протеогликаны.
Они образуются, живут 7-10 дней и распадаются под действием ферментов — лизосомных гидролаз. Последние расщепляют именно ГАГ, причем каждому виду гликозаминогликанов соответствует свой личный фермент.
Процесс образования (анаболизма) протеогликанов и распада (катаболизма) идет постоянно.
Что произойдет, если нужного фермента не окажется? Нерасщепленные или частично разрушенные протеогликаны станут накапливаться, откладываясь в соединительных тканях, пронизывающих все наше тело. Так и развиваются мупоколисахаридозы — разновидность лизосомных болезней накопления.
Какие бывают мукополисахаридозы?

На сегодняшний день известно о 8 типах заболевания, причем некоторые из них представлены в нескольких разновидностях. Обычно это связано с тем, что дефицит одного фермента может быть вызван разными мутациями — и, как следствие, проявления этих мутаций и срок жизни больных тоже могут различаться.
- I тип объединяет три фенотипа: синдром Гурлер (МПС-IH), синдром Шейе (МПС-IS) и синдром Гурлер-Шейе (МПС-IH/S). Мутации, вызывающие синдром Гурлер и синдром Шейе, происходят в одном и том же гене. И иногда они развиваются одновременно. МПС I типа считается самой распространенной разновидностью болезни. Продолжительность жизни — от 6-10 (синдром Гурлер) до 30 лет (синдром Шейе).
- II тип — синдром Хантера (МПС-II). Это второй по частоте встречаемости вид мукополисахаридозов. Продолжительность жизни — 30-40 лет.
- III тип — синдром Санфилиппо, который подразделяется на 4 формы: A (МПС-III A), B (МПС-III B), C (МПС-III C) и D (МПС-III D). Это тоже разные мутации гена, который кодирует данный лизосомный фермент. Продолжительность жизни — до 20 лет.
- IV тип — синдром Моркио. Представлен двумя формами: A (МПС-IV A) и B (МПС-IV B). Продолжительность жизни — 20-35 лет.
- VI тип — синдром Марото-Лами (МПС-VI). Продолжительность жизни — 10-20 лет.
- VII тип — синдром Слая (МПС-VII).
- VIII тип — синдром Ди Ферранте (МПС-VIII) .
- XI тип — синдром дефицита гиалуронидазы, он же — синдром Натовича (МПС-IX).
Пятым типом раньше считался синдром Шейе.
Диагноз обычно ставят в детском возрасте.
МПС и генетика

Мукополисахаридоз — это генетическое заболевание. Оно наследуется по аутосомно-рецессивному типу. Это значит, что мутация только на одной хромосомы из пары (а мы помним, что у человека 23 пары хромосом) не вызовет заболевания, потому что ген, расположенный на парной нормальной хромосоме будет исправно работать.
Благодаря этому организм будет производить нужный фермент в требуемых количествах. А вот если человек получит на обеих хромосомах одинаковые мутации в области генов, кодирующих один и тот же лизосомный фермент — у него разовьется мукополисахаридоз. Чтобы такая ситуация сложилась, оба его родителя должны быть носителями бракованного гена.
Такое совпадение случается очень редко — и поэтому мукополисахаридозы относятся к орфанным (редким) заболеваниям. Из общего списка выбивается синдром Хантера (или Гунтера, в зависимости от перевода), МПС II типа. Именно это заболевание стало первым из открытых человечеством мукополисахаридозов.
В отличие от генов, являющихся причиной других видов МПС, ген, вызывающий синдром Хантера, расположен на половой X-хромосоме. То есть его наследование сцеплено с полом, и болеют им в подавляющем большинстве случаев мальчики.
При других видах МПС, если болен один родитель, а второй здоров, ребенок получит одну хромосому с мутантным геном, а другую — такую же, но нормальную. Болезнь не разовьется.
Но половые хромосомы Х и Y отличаются друг от друга. Мальчики получают Y-хромосому от отца и X-хромосому от матери. Если мать была обладателем хотя бы одной Х-хромосомы с мутантным геном (то есть даже сама не болела, была лишь носителем), и эта «неправильная» хромосома вошла в геном ребенка, то второй здоровой хромосомы ей в пару нет. Такой мальчик заболевает.
«Люди-горгульи» и другие проявления болезни

Разные типы мукополисахаридоза имеют свои характерные особенности. Например, при МПС I, II и III типов развиваются тяжелые нарушения нервной системы, которые приводят к деменции (слабоумию). А есть и общие черты. Как мы помним, речь идет о заболеваниях, связанных с соединительной тканью. Поэтому неудивительно, что на их фоне развиваются тяжелые системные поражения костей, суставов, кожи.
При этом может меняться форма головы, а при некоторых типах МПС уже с детства формируется специфическая внешность: приплюснутая переносица и нос с крупными ноздрями, широко посаженные глаза, толстые губы и приоткрытый рот, в целом грубые выразительные черты лица.
Подметив сходство лиц таких пациентов с горгульями — каменными изображениями мифических существ характерного облика, врачи дали название данному состоянию — гаргоилизм (от французского слова gargouille).
Сегодня выделяют два основных фенотипа (внешние проявления) мукополисахаридоза:
- Гурлер-подобный фенотип. В этом случае обычно развиваются деменция, гепатоспленомегалия (увеличение печени и селезенки) и гаргоилизм.
- Моркио-подобный фенотип. У таких пациентов нормальный интеллект и не настолько выражены повреждения скелета и связок, как при Гурлер-подобном фенотипе.
Все пациенты с МПС также страдают от задержки роста (нанизм) и диспропорционального развития скелета, нарушений зрения (помутнение роговицы и др.), слуха (тугоухость), заболеваний сердца и сосудов (аритмии, гипертрофия миокарда, заболевания клапанов сердца), болезней дыхательной системы.
Специфическая терапия

Специфической называется терапия, мишенью которой являются непосредственные причины болезни. В данном случае это ферменты, которые не вырабатываются в достаточном количестве, и те нерасщепленные мукополисахариды, которые накапливаются и вызывают болезнь.
Только в середине прошлого века стали появляться препараты, позволяющие заменить недостающие ферменты. Сегодня производятся — и зарегистрированы в России — синтетические ферменты для трех типов МПС:
- МПС-I — Альдуразим (ларонидаза);
- МПС-II — Элапраза (идурсульфаза);
- МПС-VI — Наглазим (галсульфаза).
Недостаток Элапразы заключается в том, что она не проникает сквозь гематоэнцефалический барьер, то есть не попадает в мозг.
В 2015 году СМИ сообщали о начале лечения 31-летнего пациента с синдромом Хантера новым препаратом AGT-182 — комбинацией идурсульфазы и специально разработанного для этого антитела.
Предполагалось, что это позволит лекарству проникнуть в мозг. Результаты эксперимента пока не опубликованы.
Специфических лекарств для лечения других типов МПС в России пока нет. Только симптоматическая терапия, направленная на уменьшение проявлений болезни, дает возможность таким больным прожить отведенный им срок.
В 2014 году FDA одобрило препарат Вимизим (элосульфаза альфа), предназначенный для лечения МПС-IV А — разновидности синдрома Моркио. В России данный препарат не зарегистрирован.
Другой путь — субстратредуцирующая терапия, то есть угнетение выработки мукополисахаридов, избыток которых приводит к болезни. Пока такое лекарство разработано для МПС I, II и III типов (генистеин), но исследования проводились только на животных.
В июне этого года ученые из Университета Манчестера сообщили о запуске проекта, в котором участвуют дети, больные МПС.
Проект будет длиться год, и исследователи рассчитывают, что им удастся остановить разрушение центральной нервной системы при этом заболевании.
Трансплантация костного мозга
Ферментозаместительная терапия основана на введении экзогенных (то есть извне) синтетических ферментов. А трансплантация костного мозга (ТКМ) призвана заставить организм вырабатывать собственные, эндогенные ферменты при помощи пересаженных донорских стволовых клеток.
ТКМ — крайне сложная и очень дорогостоящая процедура. Она подходит далеко не всем пациентам и не со всеми типами МПС.
Лучшие результаты были получены при пересадке костного мозга детям, страдающим МПС I типа, в возрасте 2 лет, то есть до того момента, пока поражение центральной нервной системы не стало слишком обширным.
При попытках лечения методом ТКМ мукополисахаридозов II типа отмечается повышенная частота осложнений и летальность среди пациентов. Следует учитывать, что и сам процесс подбора неродственного донора возможен далеко не для всех пациентов.
Генная терапия

Одним из самых значимых направлений в области разработки методов лечения МПС является генная терапия при помощи вирусных векторов — кольцевых ДНК, способных переносить нужный ген и встраивать его в заданный участок хромосомы. И уже есть первые положительные результаты.
В июле этого года ученые из Университета Миннесоты рассказали, как закапали в нос экспериментальным мышам препарат, содержащий вирус с геном, кодирующим лизосомный фермент альфа-L-идуронидазу — именно его так не хватает больным с МПС I типа.
Введение препарата через нос позволило ему проникнуть через гематоэнцефалический барьер в мозг и защитить мозг от разрушительного действия накапливающихся мукополисахаридов.
Теперь ученые ищут возможности повторить ту же самую процедуру с участием людей.
Мукополисахаридоз в России
С 2012 года мукополисахаридоз входит в список жизнеугрожающих орфанных заболеваний и подлежит лечению по программе «7 нозологий». Если быть точными, то в список вошли только те типы МПС, от которых есть лекарства и при этом они зарегистрированы в нашей стране, то есть: МПС-I, МПС-II и МПС-VI.
Сегодня в России, по данным общественных организаций, занимающихся проблемой МПС, проживает около 200 пациентов с мукополисахаридозом. А по данным компании Санофи, в 2017 году диагноз МПС имели 98 россиян, причем 88 из них — это дети.
Сегодня лечение получают только 77 из них: 52 человека находятся на пожизненной ферментозаместительной терапии, а 25 — прошли трансплантацию костного мозга (при синдроме Гурлер эта операция может проводиться только в возрасте от 2 до 4 лет).
Из-за высокой стоимости лекарств, которые следует вводить еженедельно, расходы на это заболевание лидируют в бюджетах, выделенных на лечение орфанных болезней.
Например, для лечения людей с синдромом Хантера необходим препарат Элапраза. Стоимость одного флакона колеблется от 100 до 200 тысяч рублей. Для лечения одного пациента еженедельно требуется 300-500 тысяч рублей. И это пожизненная терапия. Должна быть.
В последние годы закупкой препаратов для пациентов с МПС занимались региональные власти. Нежелание тратить огромные деньги на отдельных пациентов с редкими болезнями приводило к бесконечным задержкам поставки лекарств — а это немедленно ухудшало состояние больных.
Но недавно, в июне стало известно, что финансирование закупок препаратов для лечения МПС с будущего, 2018 года будет производиться из федерального бюджета. Это дает надежду многим из нынешних пациентов дожить до того момента, когда их болезнь можно будет вылечить.
Мукополисахаридоз

Гликозаминогликаны – это сложные гетеросахара, которые состоят из полисахаридных цепей, состоящих из остатков сульфатированного гексозамина и глюкуроновой кислоты. Дерматансульфат, кератансульфат, гепарансульфат, хондроитин-6- и хондроитин-4-сульфат являются гликозаминогликанами.
Впервые заболевание мукополисахаридоз было описано в 1917 году Гурлером.
В настоящее время известно около 10 генетических видов мукополисахаридоза, пять из которых возникают в результате нарушения активности сульфатаз, четыре – гликозидаз и один тип развивается при нехватке трансферазы. При мукополисахаридозе происходит накопление частиц в организме, что ведет к появлению у человека различных симптомов и патологий.
Заболевание наследуется по аутосомно-рецессивному типу.
Читайте также: Конъюнктивит: причины и симптомы патологии, лечение воспаления глаз в домашних условиях
Что происходит при мукополисахаридозе?
При наличии заболевания происходит поражение системы лизосомных ферментов, которые принимают участие в катаболизме гликозаминогликанов. В результате дефицита ферментов в органах и тканях происходит накопление гликозаминогликанов, что позволяет причислить мукополисахаридоз к болезни накопления.
Накопление гликозаминогликанов приводит к нарушению функционального состояния различных систем и органов, а вследствие того, что гликозаминогликаны входят в состав соединительной ткани, одним из главных симптомов мукополисахаридоза является системное поражение скелета и задержка в физическом развитии. Особенно это выражено при I, IV, VI типах мукополисахаридоза.
Симптомы мукополисахаридоза
К симптомам мукополисахаридоза относится задержка в росте, которая начинается к концу первого года жизни ребенка.
Следует отметить грубые черты лица: большой язык, нависающий лоб, деформация зубов и ушей, гипертелоризм. Деформируется грудная клетка, ярко выражен кифоз поясничного и грудного отделов позвоночника.
Характерны гепатоспленомегалия, паховые и пупочные грыжи, ограничение подвижности суставов.
На рентгенологическом исследовании можно увидеть «рыбьи» позвонки, раннее окостенение затылочно-теменного шва. При этом формирование ядер окостенения не нарушено.

Типы мукополисахаридоза
По степени выраженности психологических симптомов и костных изменений, а также по скорости прогрессирования обменных нарушений известно 7 типов мукополисахаридоза:
- I тип, который также называют синдром Гурлера. Заболеванию характерно быстрое развитие. В моче пациентов обнаруживается большое содержание хондроитинсульфата В и гепаритинсульфата. Наследуется по аутоиммунно-рецессивному типу.
- II тип, или синдром Гунтера. При данном заболевании отмечается пигментный ретинит, тугоухость. Заболевание прогрессирует медленно. В моче также наблюдается повышенное содержание гепаритинсульфата и хондроитинсульфата В, но в меньших количествах. Наследуется по рецессивному типу, зависит от пола.
- III тип, синдром Санфилиппо. Для данного типа характерно тяжелое слабоумие и большое количество гепаритинсульфата в моче. Наследование аутоиммунно-рецессивное.
- IV тип, или синдром Моркио.Отмечена существенная деформация скелета, особенно грудной клетки. В отличие от других типов характерно отсутствие помутнения роговицы, снижения интеллекта и появления грубых черт лица.
- V тип, синдром Шейе. Характеризуется помутнением роговицы и умеренно-выраженной деформацией скелета. Наследуется по аутоиммунно-рецессивному типу.
- VI тип, или синдром Марото-Лами. Характерны отставание в росте, бочкообразная грудная клетка, грубые черты лица.
- VII тип, возникающий вследствие нехватки бета-глюкуронидазы.
Диагностика мукополисахаридоза
Диагностика заболевания основывается на проведении генеалогических, клинических и биохимических исследований.
Лечение мукополисахаридоза
Заболевание преимущественно имеет симптоматическое лечение. Пациенту назначают гормональные препараты: преднизолон, тиреоидин, АКТГ для подавления выработки мукополисахаридов. При лечении мукополисахаридозов пациенту назначают большие дозировки витамина А и препараты для нормализации сердечной деятельности. При необходимости могут назначить цитостатические средства.
Профилактика мукополисахаридоза
Специфической профилактики заболевания не существует. Необходимо проведение пренатальной диагностики для выявления нехватки ферментов в амниотических клетках.
Информация является обобщенной и предоставляется в ознакомительных целях. При первых признаках болезни обратитесь к врачу. Самолечение опасно для здоровья!
Мукополисахаридоз II типа | Мукополисахаридозы I, II, VI типов
Мукополисахаридоз II типа (синдром Хантера) cвязан с дефицитом фермента альфа-L-идуроносульфатсульфатазы, в результате чего в соединительной ткани накапливается гликозаминогликан дерматансульфат и гепарансульфат. Синдром Хантера встречается реже, чем мукополисахаридоз I, составляя приблизительно 14-15 % от всех форм мукополисахаридозов.

Тип наследования болезни Хантера рецессивный, сцепленный с Х-хромосомой. Болезнью Хантера страдают, как правило, только мальчики, однако к настоящему моменту описано 7 случаев заболевания у девочек.
МПС II типа встречается с популяционной частотой 1:140 000 – 1:156 000.
Наследование синдрома Хантера отличается от всех остальных МПС-заболеваний, так как ген, ответственный за него, – рецессивный и сцепленный с Х-хромосомой (его называют – сцепленным с полом), как и гемофилия. Девочки могут быть носителями заболевания, но, за исключением редчайших случаев, только мальчики страдают этим типом МПС.
Если женщина является носителем МПС II, то существует 50%-ный риск, что рожденный ею сын будет страдать данным заболеванием. Кроме того, существует 50%-ный риск того, что ее дочь будет носителем этого заболевания. Важно отметить, что не все женщины, родившие ребенка с МПС II, являются носителями аномального гена.
Если только один человек в семье страдает МПС II, то нельзя определить, является ли мать носителем. Однако если в семье есть и другие дети, больные синдромом Хантера, то предполагается, что именно мать является носителем заболевания.
Сестры и тети больного с синдромом Хантера по материнской линии тоже могут быть носителями и с 50%-ной вероятностью передать аномальный ген своим сыновьям.
Синдром Хантера назван в честь профессора медицины Чарльза Хантера (Манитоба, Канада), которым были впервые описаны случаи нарушений подобного типа у двух братьев в 1917 г.
Кодирование по МКБ-10 — E 76.1
Проявления МПС II
Характерны изменения черт лица по типу «гаргоилизма», которые становятся очевидными к концу первого-второго года жизни: макроцефалия, выступающие лобные бугры, запавшая переносица, короткие носовые ходы с вывернутыми кнаружи ноздрями, полуоткрытый рот, большой язык, пухлые губы.
У пациентов отмечается задержка роста, короткая шея, контрактуры суставов, грыжи, позднее прорезывание зубов, отмечаются низкий рост волос на лбу, длинные густые ресницы и брови. С возрастом волосы становятся более жесткими, прямыми и светлыми (цвета соломы).
У людей с тяжелой формой синдрома Хантера наблюдается прогрессирующая задержка умственного развития и серьезные прогрессирующие физические нарушения. Люди с легкой формой синдрома Хантера имеют нормальный уровень интеллектуального развития, легкие и медленно прогрессирующие физические нарушения, продолжительность их жизни существенно выше, чем у людей с тяжелой формой МПС II.
Большинство диагностированных пациентов с синдромом Хантера имеют нормальный или близкий к нему уровень интеллектуального развития и заметные физические отклонения, что соответствует средней степени тяжести заболевания.
На сегодняшний день нет достоверного способа, позволяющего спрогнозировать степень тяжести заболевания по результатам биохимических анализов, так как при всех формах синдрома Хантера у пациентов отсутствует один и тот же фермент.
Форму заболевания определяют на основании имеющихся симптомов и отклонений.
Детальные исследования показали, что у людей с легкой формой синдрома Хантера фермент все-таки присутствует в организме, хотя и в малом количестве, что и объясняет легкую форму течения заболевания.
Рост
Пациенты с синдромом Хантера, как правило, ниже здоровых людей, но отклонения в росте от нормы варьируют в зависимости от тяжести заболевания.
Младенцы с тяжелой формой МПС II часто рождаются довольно крупными и в течение первых двух лет растут быстрее нормы. Их рост замедляется к концу 2-го года, и в дальнейшем они растут медленнее и вырастают до 120 — 140 см.
Люди с легкой формой синдрома Хантера, как правило, имеют нормальный рост.
Кожные покровы
Для данной формы мукополисахаридоза характерно узелково-папулёзное поражение кожи, преимущественно в области лопаток, наружных и боковых поверхностей плеч и бёдер. Эти изменения обусловлены отложением липидов и гликозаминогликанов в дерме.
Костная система
Отмечается деформация кисти по типу «когтистой лапы». Характерны кифоз, деформирующий остеоартроз тазобедренных суставов, множественный дизостоз, увеличение турецкого седла.
Позвонки в нормальном состоянии выровнены по линии от шеи до ягодиц.
У людей с тяжелой формой синдрома Хантера позвоночник сформирован неправильно, один или два позвонка в средней части позвоночника иногда бывают меньше, чем остальные, и слегка сдвинуты. Такое смещение позвонков может стать причиной развития искривления позвоночника (кифоз или горб). Обычно при синдроме Хантера позвоночника искривлен несильно и не требует специального лечения.
Тугоподвижность суставов типична для всех типов мукополисахаридоза, и диапазон движений может быть ограничен во всех суставах.
С годами тугоподвижность может привести к возникновению боли в суставах, которую снимают при помощи тепла и болеутоляющих средств, типа ибупрофена, однако принимать их следует только под контролем врача, так как прием таких лекарств может вызвать раздражение желудка и язву.
Многие люди с синдромом Хантера стоят и ходят на полусогнутых ногах из-за тугоподвижности в тазобедренных и коленных суставах. Это в сочетании с напряженным ахилловым сухожилием порой заставляет их ходить на цыпочках.
Иногда встречается Х-образное искривление ног, как правило, не требующее лечения. В случае серьезных деформаций требуется операция на больших берцовых костях (при синдроме Хантера такая операция требуется крайне редко).
Ступни ног широкие и негибкие с поджатыми и искривленными, как на руках, пальцами.
Органы дыхания
Характерны частые респираторные заболевания; повторные отиты, часто приводящие к прогрессирующей потере слуха; обструктивные заболевания дыхательных путей.
Как правило, у людей с синдромом Хантера уплощенная переносица. В результате изменения костей в средней части лица и утолщения слизистой оболочки носовой проход уже, чем обычно. Такое строения носоглотки в совокупности с накоплением вещества в мягких тканях носа и горла может привести к тому, что носовые проходы легко блокируются.
Одной из отличительных черт детей с синдромом Хантера являются слизистые выделения из носа (ринорея) и хронические инфекционные заболевания ушей и пазух носа.
Миндалины и аденоиды часто увеличены и могут частично блокировать дыхательные пути. Шея обычно короткая, что способствует возникновению проблем с дыханием. Трахея часто сужается из-за накопления вещества и становится более гибкой и мягкой, чем в норме, из-за изменений в строении колец хряща в трахее.
Форма грудной клетки неправильная, соединение между ребрами и грудиной не такое гибкое, как в норме. Поэтому грудная клетка не может двигаться свободно, что, в свою очередь, не позволяет легким набирать большой объем воздуха.
Диафрагма выдвинута вверх из-за того, что внутренние органы (печень и селезенка) увеличены, что тоже уменьшает пространство, доступное легким. Если в легкие попадает жидкость, возникает высокий риск возникновения инфекции (пневмонии). При сужении дыхательных путей и повышенной секреции высока опасность возникновения астматических приступов.
Многим пациентам во время вирусной болезни уменьшить кашель и облегчить дыхание помогает применение противоастматических препаратов.
У многих пациентов, страдающих синдромом Хантера, дыхание шумное, даже когда нет никакой инфекции. Сон у них беспокойный и часто сопровождающийся храпом. Иногда у больного во сне наблюдаются остановки дыхания на короткие периоды (апноэ).
Шумное дыхание, которое останавливается и возобновляется, может порой очень напугать родителей, но такие остановки дыхания на 10-15 секунд рассматриваются медиками как норма.
Важно знать, что многие пациенты с МПС II могут дышать подобным образом в течение многих лет.
Во время остановки дыхания содержание кислорода у ребенка в крови понижается, что может спровоцировать проблемы с сердцем. Если у ребенка наблюдается существенное удушье или прерывание дыхания, необходимо проконсультироваться со специалистом, который может оценить состояние ребенка во время сна с использованием специальных тестов.
Иногда апноэ лечится при помощи удаления миндалин и аденоидов, проведения вентиляции дыхательных путей при помощи непрерывного или двухуровневого положительного давления или проведения трахеостомии (операции по рассечению передней стенки трахеи с последующим введением в ее просвет канюли либо созданием постоянного отверстия – трахеостомы).
Многие специалисты считают, что пациентам с МПС II трахеостомия показана на более ранних этапах развития болезни, чем ее обычно проводят. Пациенты после трахеостомии чувствуют себя намного лучше, так как дыхание во время сна улучшается.
Читайте также: Энцефалиты (воспаление мозга): как проявляется, методы лечения и причины
Органы зрения
Пигментная дегенерация сетчатки. Для болезни Хантера менее характерно помутнение роговицы, в отличие от МПС I и VI типов. У пациентов с тяжелой формой МПС II часто выявляется дистрофия сетчатки, приводящая к нарушению периферического и снижению сумеречного зрения. Возможен отек диска зрительного нерва, обусловленный повышением внутричерепного давления. Редко встречается глаукома.
Центральная нервная система
Задержка психомоторного и речевого развития выражена с 1,5-3 лет. При тяжелой форме заболевания к 8 годам развивается тяжелая умственная отсталость.
Отставание в развитии людей с тяжелой формой синдрома Хантера многие исследователи связывают с накоплением мукополисахаридов в нейронах головного мозга. При легкой форме синдрома Хантера отставания в интеллектуальном развитии не наблюдается.
Функции головного мозга затрагивают такие типичные при синдроме Хантера процессы, как:
- пониженный уровень кислорода (гипоксия),
- потеря сна из-за апноэ,
- повышенное давление жидкости внутри и вокруг мозга (внутричерепное давление, гидроцефалия),
- снижение зрения и слуха.
Признаки гидроцефалии зачастую появляются медленно и незаметно, и могут заключаться в изменении поведения, появлении головной боли, нарушении зрения.
Характерно наличие судорог (особенно при тяжелой форме). Симптоматическая эпилепсия развивается, как правило, при тяжелом или среднетяжелом течении заболевания. У пациентов со слабо выраженными и клиническими признаками она встречается крайне редко.
Карпальный тоннельный синдром – это частая нейропатия сдавления у пациентов в возрасте от 3 до 10 лет. В начальной стадии заболевания возникают онемение пораженной кисти, трудности выполнения тонких движений, снижение чувствительности пальцев кисти.
Эти симптомы редко отмечаются ребенком и не расцениваются родителями как патологические. Позже появляется чувство покалывания в кончиках пальцев кисти и со временем процесс может распространяться на предплечье и плечо. Пациенты редко сообщают о болевых ощущениях, пока не происходит потеря функции.
Нарушения глотания: костные изменения приводят к снижению подвижности нижней челюсти, что ограничивает способность открывать рот и жевать.
Поведенческие нарушения – гиперактивность, расторможенность, агрессивность и упрямство, как правило, имеют место у детей со среднетяжелой и тяжелой формами мукополисахаридозов.
Проблемы с поведением значительно влияют на повседневную жизнь ребенка и его социальную адаптацию.
По мере нарастания когнитивного дефицита к гиперактивности и агрессивности присоединяются аутистические черты, отмечается постепенная потеря навыков речи.
Сердечно-сосудистая система
Поражение сердца наблюдается у большинства больных. Как и при мукополисахаридозе I типа могут возникать поражение клапанов, эндомиокарда, коронарных артерий. Чаще отмечается патология митрального клапана, при этом могут быть как недостаточность, так и стеноз левого атриовентрикулярного отверстия.
Однако поражения клапанов значительно менее выражена у детей и подростков и, как правило, манифестирует у взрослых больных. Клапаны сердца могут быть повреждены накопившимися мукополисахаридами, в таком случае прослушиваются шумы сердца (звуки, вызванные быстрыми непостоянными потоками крови в сердце).
В норме клапаны сердца работают таким образом, что при прохождении крови из одной камеры сердца в другую не допускается течение крови в неправильном (обратном) направлении. Если клапан ослаблен, он закрывается неплотно, и небольшое количество крови может двинуться в обратном направлении.
Это приводит к возникновению быстрых непостоянных противотоков в общем потоке, что, в свою очередь, порождает шум. У большинства пациентов с синдромом Хантера в какой-то степени есть такое просачивание крови – порок сердца.
Зачастую у МПС-пациентов есть проблемы с аортальным или митральным клапаном, которые могут медленно прогрессировать в течение многих лет без видимых клинических проявлений. При ухудшении состояния пациента может потребоваться хирургическая операция для замены поврежденных клапанов.
При тяжелой форме синдрома Хантера из-за скоплений мукополисахаридов может быть повреждена сама сердечная мышца (кардиомиопатия). Сердце может также быть перегружено вследствие того, что приходится прокачивать кровь через измененные легкие (анормальное увеличение правой стороны сердца в результате заболевания легких или правосторонняя сердечная недостаточность).
Из-за необычных специфических проблем, которые могут быть при мукополисахаридозе, крайне желательно получать консультации кардиолога, имеющего опыт работы с МПС-пациентами.
Желудочно-кишечная система
Возможна диарея, связанная с накоплением ГАГ в нервных клетках пищеварительного тракта. С возрастом часто развиваются запоры. С ранних лет отмечается гепатоспленомегалия. Характерны пупочная и паховая грыжи.
Мукополисахаридоз
Мукополисахаридоз – генетическое заболевание, связанное с нарушением выработки организмом определённых ферментов, что способствует развитию выраженной физической и неврологической симптоматики.
Наследственные патологии всегда являются очень серьёзными, так как в большинстве случаев они не поддаются полному излечению.
Именно к таким патологиям относится мукополисахаридоз, представляющий довольно большую группу заболеваний, развивающихся из-за того, что происходит нарушение процесса ферментативного катализа гликозаминогликанов в лизосомах.
Наследуется данная патология по аутосомно-рецессивному типу — это значит, что больной такой патологией, как мукополисахаридоз ребёнок, может появиться у абсолютно здоровых родителей, каждый из которых при этом является носителем патологического гена. При этом патологией при данном типе наследования страдают как представители мужского пола, так и женского.
Наличие патологического гена в генотипе родителей и является основной причиной развития патологии – предрасполагающих факторов в этом случае не существует, поскольку генетические заболевания не связаны ни с какими условиями окружающей среды.
Разновидности
Существуют основные симптомы данного заболевания, к которым относятся:
- выраженные деформации скелета;
- деформации костей черепа, ушных раковин, искривление зубов;
- огрубение черт лица;
- задержка роста;
- часто наблюдается задержка умственного развития;
- снижение или полное ограничение подвижности суставов;
- частое развитие грыж (разной локализации);
- снижение слуха и зрения;
- поражение внутренних органов и предрасположенность к частым инфекционным заболеваниям.
В зависимости от того, насколько выраженными являются физические и психологические нарушения, выделяют семь типов такого заболевания, как мукополисахаридоз. При этом основными, которые встречаются чаще других, являются четыре типа.
Тип первый – болезнь Гурлера
Данная патология является самой быстропрогрессирующей, протекающей с выраженной симптоматикой. Характерным симптомом для неё является деформация черепа с развитием грубых черт лица.
К двум годам клиническая картина уже полностью выражена и представлена серьёзными деформациями скелета, нарушением работы сердца, печени и селезёнки, а также выявлением множественных грыж. Прогноз заболевания крайне неблагоприятен.
Тип второй – болезнь Хантера
В этом случае отмечается появление тугоухости и частое развитие инфекционных заболеваний (пневмоний, трахеитов, бронхитов). В отличие от предыдущего типа, страдают данной патологией в основном мальчики, причём болезнь протекает медленно, не вызывая серьёзных изменений костного скелета.

Дети с заболеванием мукополисахаридоз
Существует два варианта течения этого типа мукополисахаридоза – благоприятный и неблагоприятный. В первом случае пациенты с таким диагнозом могут дожить до 30 лет, во втором – умирают в подростковом возрасте. Обычно причиной смерти становятся осложнения инфекционных заболеваний.
Тип третий – синдром Санфилиппо
Характеризуется патология тяжёлой умственной отсталостью ребёнка. Причём на первых годах жизни болезнь себя не проявляет (может лишь отмечаться неуклюжая походка). Но в 3–5 лет болезнь стремительно прогрессирует, и характеризуется недержанием кала и мочи, апатичностью, задержкой роста и развитием контрактур.
Четвёртый тип – синдром Моркио
При таком заболевании до 3 лет дети развиваются нормально, но затем у них начинается отставание в развитии – замедляется рост, появляется сколиоз, отмечается вальгусная деформация стоп, огрубевают черты лица и утолщается кожный покров.
Типичными признаками данной патологии являются такие симптомы: снижение зрения, тугоухость и развитие грыж. При этом умственное развитие ребёнка не страдает. Смерть обычно наступает вследствие сердечной недостаточности из-за перенесённых ребёнком инфекционных патологий.
Есть и другие формы мукополисахаридоза. Названия этих патологий связаны с именами тех врачей и учёных, которыми впервые то или иное заболевание было описано. Встречаются эти патологии реже, чем вышеописанные 4 типа мукополисахаридоза, но тем не менее требуют подробного ознакомления с ними.
- Синдром Шейе – один из самых благоприятных вариантов течения мукополисахаридоза. При этой патологии симптомы не проявляются в раннем детстве, а лишь где-то в возрасте 6 лет. Полная картина заболевания формируется ко времени полового созревания ребёнка, при этом все изменения выражены несильно.
- Синдром Марото-Лами имеет такие типичные особенности, как отставание в росте и огрубение черт лица. Эта патология протекает либо в лёгкой, либо в тяжёлой форме.
- Когда говорят о синдроме Слая, необходимо быть точно уверенным в данном диагнозе, так как патология имеет сходство с синдромом Санфилиппо, и только с помощью биохимического исследования проводится дифференциальная диагностика.
- Синдром Ди-Ферранте по своим клиническим проявлениям схож с болезнью Моркио. Отличием является появление психомоторного и интеллектуального отставания в развитии.
Особенности лечения
Диагностика разновидности патологии проводится путём рентгенологических исследований, биохимических исследований и других методов, позволяющих установить физические и психологические нарушения.
Так как мукополисахаридоз является генетической патологией, излечение от него невозможно и прогноз болезни почти всегда неблагоприятный – вопрос лишь в том, до какого возраста сможет дожить пациент.
В то же время обязательной является коррекция нарушений, вызванных патологией, что позволяет облегчить состояние больного человека и продлить его жизнь. Именно поэтому после постановки диагноза, ребёнок должен наблюдаться у таких специалистов, как:
- ортопед;
- хирург;
- отоларинголог;
- кардиолог;
- невропатолог;
- офтальмолог;
- педиатр.
Каждый из этих специалистов способен проводить симптоматическое лечение, к примеру, ортопед корректирует нарушения, возникшие в опорно-двигательном аппарате, назначая ребёнку физиопроцедуры, такие как массаж, ЛФК и т. д.
Хирургическое лечение направлено на устранение грыж, если таковые развиваются у ребёнка с таким заболеванием, как мукополисахаридоз.
Задача отоларинголога – лечить хронические инфекции, часто возникающие при этом заболевании, контролировать слух ребёнка.
Кардиолог занимается тем, что лечит патологии сердечно-сосудистой системы, которые часто сопровождают это заболевание, а невропатолог следит за развитием неврологических нарушений, по возможности замедляя их течение.
Офтальмолог – врач, который следит за состоянием зрения больного. Ну а задача педиатра заключается в контроле общего состояния и лечения инфекционных заболеваний, которым дети с такой патологией подвержены во много раз больше, чем те, кто ею не страдает.
Конечно же, поскольку мукополисахаридоз является генетическим недугом, больной ребёнок должен наблюдаться у генетика, который может следить за течением заболевания и корректировать выраженность симптоматики.
В любом случае излечить болезнь, даже если она обнаружена на ранней стадии, невозможно, поэтому родители детей, которым был поставлен данный диагноз, должны набраться терпения и сил, чтобы облегчить ребёнку его пусть и недолгую жизнь, и сделать её максимально комфортной.
Мукополисахаридоз
Мукополисахаридоз – совокупность достаточно редких заболеваний генетического характера, возникающих в результате дефицита определенных ферментов, способствующих расщеплению отдельных видов жиров и углеводов на простые молекулы. Различного рода патологии и симптомы развиваются у человека именно вследствие накопления данных частиц в организме.
Причины
Основной причиной развития мукополисахаридоза является его наследование по аутосомно-рецессивному типу.
Патогенез
Мукополисахаридоз характеризуется поражением систем лизосомных ферментов, которые принимают непосредственное участие в катаболизме гликозаминогликанов. Данные вещества накапливаются в органах и тканях в достаточно большом количестве в результате ферментативной недостаточности. По этой причине мукополисахаридозы принято относить к заболеваниям накопления.
Таким образом, происходит нарушение работы различных органов и систем. Одним из основных проявлений мукополисахаридоза является системное поражение скелета, а также задержка физического развития (преимущественно при I-S, IV и VI типах мукополисахаридоза).
Данное обстоятельство в первую очередь обусловлено тем, что гликозаминогликаны входят в состав соединительной ткани
Читайте также: Хламидиоз у мужчин: как развивается инфекция, первые признаки и методы лечения
Симптомы мукополисахаридоза
Среди основных симптомов мукополисахаридоза можно выделить задержку роста, которая начинает проявляться, как правило, к концу первого года жизни ребенка. Кроме того среди наиболее выраженных симптомов мукополисахаридоза можно отметить грубые черты лица: большой язык, нависающий лоб, деформацию ушных раковин, гипертелоризм, искривление зубов.
Деформированная грудная клетка, а также выраженный кифоз поясничного и грудного отделов позвоночника также являются распространенными симптомами мукополисахаридоза. В большинстве случаев отмечаются гепатоспленомегалия, соответствующие ограничения подвижности суставов, паховые и пупочные грыжи.
С помощью рентгенологического исследования выявляется такой симптом мукополисахаридоза, как раннее окостенение затылочно-теменного шва. При этом нарушение ядер окостенения не наблюдается. В неврологическом статусе отмечаются такие симптомы мукополисахаридоза, как общая двигательная заторможенность, а также диффузная мышечная гипотония.
Все типы мукополисахаридоза сопровождаются ослаблением слуха в разной степени и снижением интеллекта.
В зависимости от выраженности костных изменений и нарушений психики, принято различать семь основных типов мукополисахаридоза.
- Синдром Гурлера – первый тип мукополисахаридоза, возникающий вследствие аутосомно-рецессивного наследования. Данная форма заболевания стремительно прогрессирует, а в моче пациентов отмечается повышенный уровень содержания гепаритинсульфата (гепарансульфата) и хондроитинсульфата В (дерматансульфата).
- Синдром Гунтера – второй тип мукополисахаридоза, характеризующийся развитием пигментного ретинита и тугоухости. Данная форма заболевания протекает медленно, при этом в моче пациентов отмечается содержание гепаритинсульфата и хондроитинсульфата В, но уже в меньшем объеме. Болезнь развивается в результате рецессивного наследования, сцепленного с полом.
- Синдром Санфилиппо – третий тип мукополисахаридоза, характеризующийся развитием тяжелого слабоумия и содержанием большого количества гепаритинсульфата в моче. Заболевание развивается вследствие аутосомно-рецессивного наследования.
- Синдром Моркио – четвертый тип мукополисахаридоза.
- Синдром Шейе – пятый тип.
- Синдром Марото-Лами – шестой тип.
- Седьмой тип мукополисахаридоза обусловлен нехваткой бета-глюкуронидазы.
Диагностика
Основанием для вынесения диагноза являются клинические проявления заболевания, результаты рентгенологического исследования, экскреция гликозаминогликанов с мочой, исследования активности специфических ферментов, а также амниотическая жидкость (антенатальная диагностика).

Прогноз
Все формы заболевания характеризуются неблагоприятным прогнозом. Все дело в том, что с возрастом у пациента увеличиваются изменения скелета, а также нарушения различных систем и органов.
Профилактика мукополисахаридоза
Профилактика заболевания заключается в пренатальной диагностике, основанной на непосредственном выявлении недостатка фермента в амниотических клетках.
Мукополисахаридоз
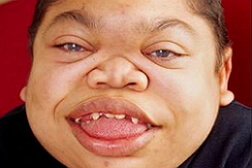
Мукополисахаридоз – это комплекс генетически детерминированных патологий, имеющий единые патогенетические механизмы развития, заключающиеся в неполном разрушении и накоплении мукополисахаридов.
Клиническая картина мукополисахаридоза формируется в зависимости от поражения структуры, в которой накапливается избыточное количество глюкозаминогликанов. Наиболее часто встречается мукополисахаридоз у детей и научно доказанным является факт передачи патологического гена по наследству.
Данная патология является чрезвычайно редкой, однако внимание исследователей приковано к проблеме поиска эффективного метода ее лечения, так как уровень летальности от мукополисахаридоза очень высок.
Причины мукополисахаридозов
Все формы и типы мукополисахаридозов относятся к категории наследственных патологий, передающихся аутосомно-рецессивным типом наследования. Мутационный ген представляет собой изменение структуры гена лизосомной альфа-ирунидазы, которая принимает непосредственное участие в метаболических превращениях глюкозаминогликанов.
Вследствие мутационного поражения лизосомной альфа-ирунидазы, происходит нарушение процесса внутрилизосомного распада дерматансульфата и избыточное его накопление в печеночной и селезеночной паренхиме, хрящевой ткани и надкостнице, нервных тканях и сосудистой стенке.
В результате прогрессирования отека мягкой оболочки мозга развивается частичная окклюзия субарахноидального пространства, способствующая в свою очередь прогрессированию гидроцефалии. Причиной возникновения у ребенка признаков умственной отсталости является избыточное скопление ганглиозидов в нейронах.
Помимо выраженных нарушений метаболизма мукополисахаридов наблюдаются признаки обменных нарушений белков, которые проявляются в виде гипераминоацидурии.
Симптомы мукополисахаридозов
Все пациенты, страдающие мукополисахаридозом, имеют характерные отличительные фенотипические признаки в виде развития уродливых черт лица с увеличенным языком и большой головой.
Больные любым типом мукополисахаридоза имеют признаки отставания в физикометрическом развитии, а именно несоответствие костного возраста паспортному, развитие выраженной степени сколиоза и обезображивающих контрактур крупных суставов.
Несмотря на преимущественное поражение нервной и/или опорно-двигательной системы, при некоторых типах мукополисахаридоза отмечается повреждение внутренних органов.
Характерными отличительными симптомами мукополисахаридоза считается поражение нейронов корковых структур головного мозга, вследствие чего у ребенка развиваются признаки нарушения интеллектуальной способности различной степени интенсивности.
При прогрессивном течении заболевания практически в 100% случаев мукополисахаридоза отмечается поражение зрительного нерва, а также роговицы одного/обоих глаз, сопровождающееся выраженным нарушением зрительной функции.
Типы мукополисахаридозов
Самой тяжелой формой данной патологии считается мукополисахаридоз 1 типа, сопровождающийся глубокими нарушениями неврологического статуса ребенка и комплексным поражением практически всех жизненно-важных органов и систем.
Дебют заболевания приходится на грудной период и сопровождается выраженными нарушениями формы и размеров мозгового черепа, кифосколиотической деформацией грудного отдела позвоночника и контрактурами мелких суставов. У данной категории пациентов отмечается врожденное увеличение размеров печени и слабость сухожильно-мышечного пахового кольца с появлением признаков паховых грыж.
На ранних стадиях заболевания диагноз «мукополисахаридоз» редко устанавливается в связи с тем, что данная патология относится к категории редко встречающихся. Однако прогрессирование нарушений физического и умственного развития у ребенка в сочетании с внешними проявлениями позволяет ставить мукополисахаридоз 1 типа в дифференциально-диагностический ряд.
Неврологический статус ребенка, страдающего мукополисахаридозом 1 типа, имеет характерные особенности в виде развития признаков гипертензивно-гидроцефального симптомокомплекса, а именно увеличения размеров мозгового отдела черепа, появления расширенной венозной сети в височных областях и вегетативными дисфункциями.
При выполнении рентгенографии черепа в боковой проекции обращает на себя внимание некоторое увеличение саггитального размера турецкого седла, а люмбальная пункция позволяет определить повышенное ликворное давление.
Патогенез развития неврологических нарушений у пациентов с мукополисахаридозом 1 типа основан на изменениях структуры костной ткани в проекции черепа и повышенной гидрофильности соединительнотканного компонента мозговой ткани.
Поражение внутренних органов при мукополисахаридозе 1 типа происходит в отдаленном периоде и заключается в формировании пороков клапанного аппарата сердца, помутнения роговицы глаза.
В связи с тем, что данная патология сопровождается выраженными нарушениями структур головного мозга, ее относят к категории неблагоприятных в отношении жизни ребенка, длительность которой не превышает десятилетний рубеж.
Лабораторные хроматографические исследования мочи детей, страдающих этим типом мукополисахаридоза, сопровождаются определением высоких показателей уровня глюкозаминов превышающие рубеж 300 мг в сутки.
Мукополисахаридоз 2 типа имеет сходные с предыдущей формой клинические проявления, однако характерной особенностью его является преимущественное поражение лиц мужского пола на первом году жизни.
Фенотипические проявления этой патологии менее выражены по сравнению с мукополисахаридозом 1 типа, но подчеркивают генетическое происхождение заболевания: отставание в росте, гипертелоризм и уплощение переносицы, укорочение шейного отдела позвоночника и большие размеры языка.
Главным отличительным признаком 2 типа мукополисахаридоза является полное отсутствие признаков статической деформации позвоночника в виде развития кифосколиоза.
Нарушение интеллектуально-мнестической функции коры головного мозга имеет меньшие проявления, однако в большинстве случаев у детей формируются признаки сенсомоторной тугоухости.
Поражение глаз заключается лишь в небольших застойных изменениях зрительного нерва и двухстороннем помутнении поверхностных слоев роговицы.
Для 2 типа мукополисахаридоза характерно проявление эндокринных и вегетативно-трофических изменений в виде развития абдоминального типа ожирения, гипертрихоза и акроцианоза.
Дети, страдающие мукополисахаридозом 2 типа, относятся к категории риска по возникновению респираторных вирусных заболеваний, осложняющихся бактериальными пневмониями.
Диагностическим критерием установления диагноза мукополисахаридоз в данной ситуации является увеличение хроматографических показателей гепарансульфата или дерматансульфата в моче.
Синдром Санифилиппо, или мукополисахаридоз 3 типа, одинаково поражает детей мужского и женского пола и сопровождается развитием специфической фенотипической внешности.
Дети, страдающие 3 типом мукополисахаридоза, имеют аномалии развития мозгового и лицевого отделов черепа в виде формирования увеличенных лобных костей, уплощенной переносицы, большого языка и толстых губ, а также близко расположенных глазных орбит.
Поражение костно-суставного аппарата больше затрагивает мелкие суставы, нежели позвоночник, и проявляется в виде развития врожденных контрактур межфаланговых и плюснефаланговых суставов рук и ног. Также для этого врожденного синдрома не характерно поражение внутренних органов, однако неврологический статус ребенка страдает в значительной степени.
С раннего возраста дети имеют склонность к повышенной раздражительности и эмоциональной неустойчивости, нарушению памяти и прогрессирующим торможением интеллектуальной активности.
Диагноз, как правило, устанавливается на поздней стадии заболевания с помощью привлечения лабораторно-инструментальных методик обследования (рентгеноморфометрия, хроматографическое определение кислых мукополисахаридов, характеризующееся высоким показателем уровня гепарансульфата).
4 тип мукополисахаридоза, или синдром Моркио, отличается от предыдущих форм благоприятным течением, так как данная патология не сопровождается грубыми неврологическими дефектами и в большей степени затрагивает развитие костно-суставной системы ребенка.
Дети с синдромом Моркио имеют грубые аномалии развития крупных трубчатых и плоских костей, а также деформации суставов с выраженным нарушением их функции.
Лабораторное исследование мочи ребенка, страдающего мукополисахаридозом 4 типа, сопровождается обнаружением повышенного содержания дерматансульфата.
Синдром Шейе, или мукополисахаридоз 5 типа, имеет клинико-лабораторную симптоматику сходную с первым типом, однако при данной патологии отмечается более агрессивное течение заболевания с полным отсутствием периодов ремиссии.
В связи с этим, синдром Шейе занимает лидирующее положение в структуре младенческой смертности от мукополисахаридоза.
Принципиальным отличием этого синдрома является преимущественное поражение сердечно-сосудистой системы с формированием грубых пороков развития клапанного аппарата сердца и крупных сосудов.
Дети с диагностированным мукополисахаридозом 6 типа могут длительное время жить с данным заболеванием, так как данная патология не сопровождается нарушением интеллектуально-мнестических функций головного мозга и проявляется лишь в некотором помутнении роговицы, имеющем односторонний или двухсторонний характер.
Хроматографическое исследование мочи, которое входит в алгоритм обследования пациентов с подозрением на мукополисахаридоз, позволяет определить повышенный уровень дерматансульфата при отсутствии гепарансульфата.
Лечение мукополисахаридоза
Важное значение в лечении ребенка с мукополисахаридозом имеет раннее установление диагноза с определением клинико-лабораторного типа данной патологии, так как каждый вариант течения заболевания требует индивидуального подхода к применению целесообразного метода лечения.
В определении тактики ведения и лечения пациента, страдающего мукополисахаридозом того или иного типа принимает участие группа специалистов, включающая педиатра-неонатолога, специалиста пластической хирургии, ортопеда, нейрохирурга, офтальмолога и отоларинголога.
Как правило, лечение мукополисахаридоза заключается в устранении тех или иных признаков заболевания, то есть применяется симптоматический вариант терапии.
Единственным патогенетически обоснованным методом лечения в настоящий момент считается гормонотерапия, позволяющая не только купировать клинические проявления, но и нивелировать лабораторные проявления заболевания.
Оптимальной комбинацией, позволяющей значительно снизить активность синтеза глюкозаминогликанов фибробластами, является сочетание Тиреоидина и Преднизолона (суточная дозировка составляет не менее 1 мг на 1 кг веса ребенка курсом 2 месяца).
Целесообразно также назначение препаратов, улучшающих функцию фибробластов, к которым относится Метионин в суточной дозе 0,75 мг перорально.
Мукополисахаридоз 1 типа является абсолютным показанием для проведения трансплантации костного мозга, при условии отсутствия противопоказаний, причем большей эффективностью этот метод обладает в возрасте пациента до двух лет.
Имеющиеся экстраневральные нарушения у ребенка являются обоснованием для применения заместительной терапии с применением Альдуразима в дозировке 100 ЕД на кг веса ребенка методом еженедельной внутривенно-капельной инфузии.
Однако данное лекарственное средство не применяется в лечении мукополисахаридоза 2 типа, так как действующее вещество неспособно проникнуть через гематоэнцефалический барьер.
Похожие записи:
- Гормональные контрацептивы пролонгированного действия: описание средств и нюансы их использования
- Желчный перитонит: причины развития, клинические формы, сопутствующие симптомы и способы лечения
- Околочелюстной абсцесс: механизм развития, сопутствующие симптомы, способы лечения и возможные осложнения
- Ребенок кусается в 1-2 года в детском саду — как отучить?
Источник https://www.krasotaimedicina.ru/diseases/traumatology/mucopolysaccharidosis
Источник https://probolezny.ru/mukopolisaharidoz/
Источник https://stavkdp.ru/articles/hirurgiya/mukopolisaharidoz-1-2-tipa-faktory-riska-razvitiya-harakternye-simptomy-osobennosti-lecheniya-i-prognoz.html