Причины, симптомы и лечение патологии Гентингтона, склонность к болезни
Болезнь Гентингтона (код по МКБ-10 – G10) – это очень редкое наследственное нейродегенеративное заболевание, вызываемое нарушениями клеток в мозге (в стриатуме) с их последующим отмиранием и поражением ЦНС.
Описание
Хорея Гентингтона – что за болезнь, в чем ее суть? Это серьезное наследственное заболевание с типичными психическими и физическими симптомами, которые обычно возникают между 30 и 45 годами. Но признаки могут появиться раньше. Если они наблюдаются в возрасте около 20 лет, патология упоминается как ювенильная болезнь Гентингтона (вариант Вестфаля). Как правило, она характеризуется неконтролируемым подергиванием конечностей, туловища, головокружением. Заболевание приводит к психологическому расстройству личности, слабоумию.
Синдром Гентингтона затрагивает около 5-10 человек на 100000. К сожалению, ее невозможно предотвратить, замедлить или вылечить. Ожидаемая продолжительность жизни уменьшается не самой болезнью, а ослабленным иммунитетом.
Патология получила свое название в честь американского врача Джорджа Гентингтона, впервые описавшего ее в 1872 г.
Причины и факторы риска
Причина заболевания – изменение (мутация) гена IT15 на 4-й хромосоме.
Основой мутации при болезни Гентингтона является размножение триплета CAG (цитозин-аденин-гуанин). Нормальное состояние – до 35 триплетов CAG, при количестве 36-39 прогноз неясен, при более 40 триплетах человек наверняка заболеет этой коварной болезнью. Правило «чем больше триплетов CAG, тем быстрее вспышка заболевания» особенно верно для людей с экстремальным количеством триплетов CAG.
Ген, содержащий менее 36 триплетов, продуцирует белок хантингтин, необходимый для правильного развития мозга. Болезнь Гентингтона – это заболевание с аутосомно-доминантным типом наследования. Это означает, что она встречается в основном в каждом поколении. Если один из родителей страдает этим расстройством, существует 50% риск того, что ребенок также заболеет.

При диагностике болезни Гентингтона необходимо учитывать, что не всегда известно присутствие в семье носителя мутаций. Причины – преждевременная смерть от другой патологии, развод, неизвестность отца. Новые мутации встречаются очень редко.
Пациентам, страдающим от заболевания, рекомендуется не иметь детей. Но проблема заключается в том, что в большинстве случаев оно диагностируется только после рождения ребенка. Генетическая мутация присутствует с рождения, но обычно начинает проявляться во взрослом возрасте.
Симптомы
Человек обычно приходит к врачу из-за проблем с движением, сопровождающихся изменением психики. Симптомы и признаки болезни Гентингтона чаще всего появляются около 40 лет. Клиническая картина постепенно ухудшается в результате повреждения головного мозга.
Вначале наблюдаются мелкие мышечные подергивания и постоянные движения конечностей. Позже развивается полная хорея. Она сначала поражает руки и голову, человек делает различные гримасы, открывает рот. Затем затрагиваются нижние конечности. Эти непроизвольные движения значительно усиливаются при стрессе, в то же время, вообще не появляются во время сна. Движения прогрессируют, человек становится неспособным к самостоятельности. Признаки проявляются на обеих сторонах тела.
Типичный симптом болезни хорея Гентингтона – нарушение походки, которая иногда напоминает танец. На более поздней стадии происходят падения, неуверенные движения. Непроизвольные движения могут больше не проявляться, развивается ригидность, человеку трудно двигаться.
Заболевание часто сопровождается нарушением речи (она становится непонятной). Кроме того, наблюдается расстройство глотания, приводящее к серьезным проблемам с приемом пищи. Пищевые расстройства вызывают потерю веса.
В большинстве случаев при болезни Гентингтона возникает недержание мочи.
Значимые проявления генетического заболевания хорея Гентингтона – психические проблемы. Первоначально возникает незначительная раздражительность, затем появляются другие признаки, такие как:
- агрессивность;
- апатия;
- безжалостное поведение;
- склонность к воровству;
- сниженный интерес к внешности;
- общие изменения личности.
Хотя окружающие замечают эти проблемы, немногие приписывают их болезни.
Депрессия возникает уже на ранних стадиях заболевания. Появляется страх перед новым днем, опасения потери производительности, неудач, нехватка энергии, воли к действию. Человек становится безразличным к окружающему миру, раздражается, имеет трудности с чувством радости, не концентрируется, страдает нарушением памяти. В результате психических проблем возникает снижение аппетита, потеря веса, нарушения сна. Больного могут даже посещать мысли о самоубийстве. Частью депрессии бывает тревожное расстройство.
Депрессия может чередоваться с периодом гипомании, которому присуще чувство полноты энергии, активности.

Больному не хватает проницательности и самокритики, поэтому ему трудно понять, что он больше не может продолжать принимать решения о содержании семьи или управлять автомобилем.
Серьезные проблемы включают галлюцинации и бред. Галлюцинация – это расстройство восприятия, при котором пациент не может провести различие между реальностью и картинами, которые рисует его воображение. Он воспринимает звуковые и визуальные галлюцинации как истинные, попытки переубеждения бессмысленны. Бред – это расстройство мышления, при котором больные страдают манией преследования, считают, что имеют особое происхождение и способности, бессмертность, безнаказанность и т.д.
Основные клинические симптомы болезни Гентингтона включают слабоумие, встречающееся у всех пациентов. Человек перестает выполнять обычную работу, не может сосредоточиться, постоянно забывает.
Ограниченная подвижность и слабоумие – одни из самых печальных проявлений, потому что они лишают человека самостоятельности на всю жизнь.
Выделяются 3 основных типа болезни Гентингтона:
- Ювенильная форма. Появляется к 20 годам. При этом типе непроизвольные движения обычно отсутствуют, но возникает мышечная скованность. Наблюдается быстрое развитие деменции, расстройств личности, речевых нарушений.
- Классическая форма. Проявляется в возрасте 35-50 лет и встречается в 90% случаев. Характеризуется типичным курсом, описанным выше.
- Форма позднего старта. Появляется после 60 лет. Из всех трех типов этот является одним из самых легких – его течение медленное, значительное слабоумие отсутствует, человек сохраняет самостоятельность.
Диагностика
Фундаментальное обследование – генетическое, доказывающее умножение триплетов CAG. В соответствии с уровнем мутаций оно определяет приблизительный возраст, в котором может проявиться расстройство. Такое тестирование предполагает соблюдение очень строгих этических правил. Только пациенты, считающие это целесообразным, полностью согласные с тестом, могут сдавать анализы. Совершенно недопустимо принуждать человека к тестированию, переубеждать его. Из-за невозможности лечения необходимо связать эти тесты с поддержкой психолога и, при необходимости, социальных работников.

Из вспомогательных исследований (а также в рамках дифференциальной диагностики) можно выполнить КТ или МРТ. Цель этих обследований – определение нарушения базальных ганглиев, особенно хвостатого ядра.
У пациентов с подозрением на болезнь Гентингтона проводится специальный тест. Он подтверждает или исключает подозрение со 100% достоверностью. Всегда необходима подпись пациента, фиксирующая ознакомление с правилами диагностики.
Предиктивный (предсимптомный и пренатальный) тест проводится у бессимптомных людей с заболеванием в семейной истории, желающих узнать, поражены ли они болезнью. Но генетический тест только подтверждает наличие мутации и предпосылок к болезни, а не сам диагноз.
При беременности у женщин, у которых развилась хорея Гентингтона, диагностика может заключаться в проведении теста, направленного на определение генетического статуса их будущего ребенка. Этот пренатальный тест выполняется путем отбора амниотической жидкости (амниоцентез).
Лица, желающие пройти предсимптомное тестирование, должны тщательно рассмотреть последствия. Они имеют право не знать свой генетический статус.
В связи с ответственностью, предсимптомное тестирование не проводится для лиц младше 18 лет.
Лечение
Болезнь Гентингтона в настоящее время неизлечима. Врачи пытаются противодействовать многим ее проявлениям. Заболевание требует сотрудничества специалистов из области неврологии, психологии, психиатрии, физиотерапии, трудотерапии, логопедии и др.
В очень серьезных случаях непроизвольных движений используются антипсихотические, успокоительные средства или нейролептики. Они также применяются в случаях бреда, галлюцинаций, агрессии, беспокойства. Однако такие лекарства имеют много побочных эффектов, поэтому находят применение только в действительно тяжелых случаях, после тщательной оценки возможных рисков.

В процессе лечения хореи Гентингтона важно бороться с потерей веса, принимая высококалорийную пищу (более 5000 ккал/день). Желательно обсудить питание с диетологом. Рекомендуется употреблять жирное мясо, цельные молочные продукты, жирную рыбу, сладкие соки, картофель, шоколад.
Против депрессии эффективны антидепрессанты. При нарушениях сна, вызванных беспокойством, по рекомендации врача принимаются снотворные препараты. Возможно также применение народных методов, например, успокаивающих травяных чаев.
К сожалению, самое серьезное проявление заболевания – слабоумие – совершенно неизлечимо. Его даже нельзя замедлить.
Осложнения
Одно из осложнений – инфекции. Наиболее распространенная инфекция – пневмония, способная быть смертельной для пациента из-за ослабленного иммунитета.
Сосудистые нарушения, закупорка артерий могут привести к сердечной недостаточности. Серьезное осложнение – расстройство глотания и приема пищи. Пациент сильно худеет, истощается.

Все остальные осложнения связаны с симптомами, относящимися к самой болезни. Особенно разрушительным является изменение в поведении. Часто человек становится эгоцентричным, агрессивным, навязчивым. Осложнение этого поведения – более высокая склонность к зависимости. Опасность представляют тенденции к самоубийству, которые при болезни Гентингтона намного выше, чем в среднем по населению.
Важно знать
Болезнь Гентингтона связана не только с самим пациентом, но и с его семьей, окружением. Самые большие опасения относятся к здоровью потомства – генетический риск очень высок. Также нелегко объяснить детям, что на самом деле происходит с их родителями. Почему мама ведет себя так странно, что делает со своими руками и т.д. Часто тема заболевания становится запретной в широкой семье или в обществе, что усугубляет психическое состояние пациента.
Жизнь партнера больного человека выворачивается наизнанку. Он постепенно теряет свое свободное время, которое должен посвятить, заботе о нем. У большинства людей, живущих в семье с больным, развивается депрессия, негативное отношение к миру, вспышки гнева, постоянная грусть и т.д. Нередки случаи пристрастия к алкоголю.
К сожалению, в нашей стране нет специализированного учреждения долгосрочного или краткосрочного лечения этого заболевания. В то же время социальная поддержка играет жизненно важную роль в оказании помощи пациентам и их семьям.
Лечение хореи Гентингтона (заболевание)

Распространенность генетического заболевания хореи Гентингтона составляет порядка 10 случаев на 100 тысяч человек в мире. Выраженными признаками при диагнозе становятся нарушения в работе опорно-двигательного аппарата, судороги, резкие неосознанные рывки конечностей и ухудшение сознания. Лечение хореи Гентингтона (заболевание по типу нарушения белковой структуры генов) еще не создано, можно только медикаментозно уменьшать симптомы больного, а также способствовать сокращению двигательных расстройств.
Хорея Гентингтона, что это за болезнь
Хорея Гентингтона относится по своей типологии к нейродегенеративной болезни, во время которой у человека происходит удлинение повторов гена, который отвечает за белок гентингтин. До сих пор ученые не смогли однозначно установить функцию, выполняемую белком. Однако, в медицине существуют определенные нормы, которые указывают на длину триплетов (генов) у здорового человека.
Хорея Гентингтона, что это за болезнь с точки зрения сложной генетики человека? У здоровых людей количество повторений белка гентингтина колеблется в зависимости от возраста и других особенностей в организме в пределах 11 – 34 триплетов. При диагнозе хореи Гентингтона количество повторений начинается с показателя 37 и может достигать 100 триплетов. Этот дефект генов начинает проявляться не с рождения, а только после достижения человеком возраста 30-40 лет.
Заболевание хорея Гентингтона
Учеными доказано, что более чем у 90% пациентов во всем мире заболевание хорея Гентингтона диагностируется в возрасте 40-50 лет. Именно в этот период жизни у больного начинают активно проявляться все симптомы и признаки генного заболевания, которые невозможно остановить. В настоящее время такая болезнь считается неизлечимой, поскольку проявляется на фоне нарушения генетического строения головного мозга. Лечение хореи построено на принципе облегчения жизни пациенту, и уменьшения проявления симптомов, связанных с нарушением функций опорно-двигательного аппарата.
Во всем мире заболевание имеет разную продолжительность периода дожития человека после появления диагноза. Средний срок продолжительности жизни больного хореей Гентингтона составляет 15 лет. Такой длительный срок наблюдается лишь у людей, получающих постоянное консервативное лечение. Не более 7-9 лет проживают те больные, которые не получают должного лечения и не наблюдаются у специалистов.
Симптомы хореи Гентингтона
Чаще всего начало симптоматики проявляется у взрослых людей, достигших 30 летнего возраста, но всегда есть исключения и болезнь может начать развиваться раньше. Все симптомы проявляются слабо в самом начале заболевания и начинают прогрессировать со временем. Основные симптомы хореи Гентингтона выглядят следующим образом:
- Очень сложно диагностировать специфическое заболевание простому человеку или родственнику больного, поскольку на протяжении первых 6-12 месяцев хорея может просто напоминать повышенную суетливость человека или неусидчивость.
- После развития первых симптомов постепенно начинает возрастать интенсивность нарушений в работе опорно-двигательного аппарата.
- Даже при спокойном состоянии и без физических нагрузок человек может испытывать внезапные судороги верхних и нижних конечностей тела. Одним из симптомов становятся спазмы мышечной мускулатуры лица, которые проявляются при разговоре или выглядят, как гримасничество. Со временем начинает проявляться усиленная жестикуляция, становящаяся не к месту. Внимание и мышление становится рассеянным, спутанным и затруднённым.
- На прогрессирующей стадии развития болезни к симптомам добавляется неуверенная походка, напоминающая человека в странном танце. Это происходит из-за нарушения координации опорно-двигательного аппарата при нагрузках.
Признаки хореи Гентингтона
- признаки в нарушении познавательной функции, которые чаще всего затрагивают ориентацию в пространстве и зрительного осязания предметов. Появляющиеся проблемы мешают больному человеку нормально видеть изображение или картину, происходящую вокруг него. С каждым днем все сложнее становится распознать какие-либо схематичные рисунка, элементы на экране телевизора или буквы в газете;
- признаки, связанные с нарушением сознания или нарушение интеллектуальной функции больного. Появляются сбои в работе памяти, но это сказывается не на способности помнить информацию из прошлого, а на работоспособность мозга. Человек уже не может, как прежде, заниматься продуктивно несколькими делами. Появляются затруднения больного при планировании своего времени либо организации деятельности;
- признаки, проявляющиеся в нарушении эмоциональных и поведенческих функций. За больным могут замечать резкие порывы агрессии или раздражительности, которые не были ни чем вызваны. Практически половина пациентов страдает от депрессии, но сама этого не осознает.
Диагностика хореи Гентингтона
Если человек жалуется на вышеперечисленные симптомы, а родственники или близкие наблюдают у больного развитие всех признаков болезни, то диагностика хореи Гентингтона в первую очередь начинается с похода к врачу и назначения пациенту генетического исследования.
Диагностика позволит выявить у человека паталогический ген, который привел к развитию хореи Гентингтона. Только в этом случае можно говорить о подтверждении данного диагноза. Кроме генетического исследования, в качестве видов диагностики заболевания в медицинской практике применяют компьютерную и магнитно-резонансную томографию коры головного мозга.
Сложное генетическое исследование состоит анализа ДНК, по результатам которого врач может судить о наличии у человека аномального гена. Заболевание может передаваться по наследству, поскольку с вероятностью более 50% повторные гены передаются детям от одного из больных родителей.
Лечение хореи Гентингтона
Болезнь заключается в генетическом отклонении человека, поэтому в настоящее время не существует её специфического лечения. Все силы врачей могут быть направлены на поддержание психического равновесия человека, а также на устранение двигательных и поведенческих расстройств. Благодаря специальным медикаментозным препаратам, удается свести к минимуму проблемы с походкой человека или его нахождением в обществе.
Для лечения и предупреждения развития болезни хореи Гентингтона используют комплексное лечение, но только после проведенного анализа дезоксирибонуклеиновой кислоты и медицинского генетического консультирования. Мутантный ген не поддается лечению, и важно на начальных этапах развития болезни не спутать её с шизофренией, наследственной атаксией или же болезнью Альцгеймера. Именно для этого проводят анализ ДНК, поскольку при иных психических расстройствах число повторов тринуклеотидов не увеличивается.
Лекарство для лечения хореи Гентингтона
- Фенотозианы способствуют блокировке дофаминовых рецепторов в организме человека. Большинство препаратов из этой группы, например, Прометазин или Фторфеназин, заглушают работу гистаминовых и серотониновых рецепторов человека.
- Галоперидоловая группа препаратов направлена на уменьшение расстройств поведения. Препараты оказывают нейролептическое свойство, помогают заблокировать дофаминовые рецепторы в организме человека и частично восстановить поведенческие функции. Однако, на фоне приема сильнодействующих средств у больного очень часто развиваются такие побочные эффекты, как постоянная сонливость, проявление резких двигательных движений, не поддающихся самоконтролю больного и снижение мышечной мускулатуры всего тела.
- Группа бензодиазепинов в составе лекарственных препаратов назначается пациентам для уменьшения судорожных реакций организма. Кроме того, препараты на основе бензодиазепинов способствуют более хорошему сну пациента. Эффект от таких лекарств достигается за счет торможения рецепторов ГАМК (аминомасляной кислоты), которые стимулируются работой нервной системы.
Клиника лечения хореи Гентингтона
Лечение заболевания должно проводиться только под руководством и присмотром врачей психиатрической клиники. Генетически сложная болезнь в любое время может проявляться по-разному, поэтому важно не упустить момент начала появления симптомов. Клиника лечения хореи Гентингтона должна специализироваться на неврологических отклонениях у людей.
Медленно прогрессирующее заболевание нервной системы и коры головного мозга неизлечимо, но человек с таким диагнозом должен постоянно находиться под наблюдением специалиста, который сможет в экстренной ситуации правильно рассчитать и применить медицинский препарат. За больными людьми необходим бережный уход, а также присмотр таких врачей, как психиатры, неврологи, психологи, окулисты и ортопеды.
Клинические рекомендации при хорее Гентингтона
Список основных рекомендаций должен составлять индивидуально после осмотра больного человека психиатром и неврологом. Выделяются основные клинические рекомендации при хорее Гентингтона, которым необходимо придерживаться во время нахождения в стационаре или при нахождении пациента дома. Больной должен регулярно получать препараты, снижающие активность дофаминов коры головного мозга, доза при этом увеличивается каждые 3 дня.
Родителям, у одного из которых выявлено генетическое заболевание, не рекомендуется заводить детей. Вероятность наследования хореи Гентингтона превышает 50%, при наступлении беременности требуется провести анализ ДНК будущему ребенку. По статистике, люди с хореей Гентингтона чаще всего умирают от пневмонии, сердечной деятельности или других застойных процессов в организме, поэтому важно своевременно проводить лечение любых второстепенных заболеваний.
Круглосуточные бесплатные консультации
Мы будем рады ответить на все Ваши вопросы!
Частная клиника «Спасение» уже 19 лет осуществляет эффективное лечение различных психиатрических заболеваний и расстройств. Психиатрия – сложная область медицины, требующая от врачей максимума знаний и умений. Поэтому все сотрудники нашей клиники – высокопрофессиональные, квалифицированные и опытные специалисты.
Когда обращаться за помощью?
Вы заметили, что Ваш родственник (бабушка, дедушка, мама или папа) не помнит элементарных вещей, забывает даты, названия предметов или даже не узнает людей? Это явно указывает на некое расстройство психики или психическое заболевание. Самолечение в таком случае не эффективно и даже опасно. Таблетки и лекарства, принимаемые самостоятельно, без назначения врача, в лучшем случае на время облегчат состояние больного и снимут симптомы. В худшем – нанесут здоровью человека непоправимый вред и приведут к необратимым последствиям. Народное лечение на дому также не способно принести желаемых результатов, ни одно народное средство не поможет при психических заболеваниях. Прибегнув к ним, Вы лишь потеряете драгоценное время, которое так важно, когда у человека нарушение психики.
Если у Вашего родственника плохая память, полная потеря памяти, иные признаки, явно указывающие на психическое расстройство или тяжелую болезнь – не медлите, обращайтесь в частную психиатрическую клинику «Спасение».
Почему выбирают именно нас?
В клинике «Спасение» успешно лечатся страхи, фобии, стресс, расстройство памяти, психопатия. Мы оказываем помощь при онкологии, осуществляем уход за больными после инсульта, стационарное лечение пожилых, престарелых пациентов, лечение рака. Не отказываемся от больного, даже если у него последняя стадия заболевания.
Многие государственные учреждения не желают браться за пациентов в возрасте за 50-60 лет. Мы помогаем каждому обратившемуся и охотно осуществляем лечение после 50-60-70 лет. Для этого у нас есть все необходимое:
- пансионат;
- дом престарелых;
- лежачий хоспис;
- профессиональные сиделки;
- санаторий.
Старческий возраст – не причина пускать болезнь на самотек! Комплексная терапия и реабилитация дает все шансы на восстановление основных физических и мыслительных функций у подавляющего большинства пациентов и значительно увеличивает продолжительность жизни.
Наши специалисты применяют в работе современные способы диагностики и лечения, самые эффективные и безопасные лекарственные препараты, гипноз. При необходимости осуществляется выезд на дом, где врачами:
- проводится первичный осмотр;
- выясняются причины психического расстройства;
- ставится предварительный диагноз;
- снимается острый приступ или похмельный синдром;
- в тяжелых случаях возможно принудительное помещение больного в стационар – реабилитационный центр закрытого типа.
Лечение в нашей клинике стоит недорого. Первая консультация проводится бесплатно. Цены на все услуги полностью открытые, в них заранее включена стоимость всех процедур.
Родственники больных часто обращаются с вопросами: «Подскажите что такое психическое расстройство?», «Посоветуйте как помочь человеку с тяжелой болезнью?», «Сколько с ней живут и как продлить отведенное время?» Подробную консультацию Вы получите в частной клинике «Спасение»!
Мы оказываем реальную помощь и успешно лечим любые психические заболевания!
Проконсультируйтесь у специалиста!
Мы будем рады ответить на все Ваши вопросы!
Как спасти Тринадцатую? (Перспективы лечения болезни Хантингтона)
Статья на конкурс «био/мол/текст»: Сейчас сложно найти человека, который никогда не слышал про болезни Альцгеймера, Паркинсона или Хантингтона. Эти недуги относятся к группе нейродегенеративных заболеваний, вызывающих гибель нейронов и постепенное разрушение головного мозга. К сожалению, все они являются неизлечимыми. Поэтому ученые активно работают над тем, чтобы раскрыть механизмы развития этих болезней и найти терапию, которая поможет спасти пациентов. В своем исследовании мы обратились к пока еще малоизученному вопросу — что происходит с синаптической связью нейронов при нейродегенеративном процессе? Результаты этой работы открывают новое направление для разработки лекарства от болезни Хантингтона и других нейродегенеративных заболеваний.

Конкурс «био/мол/текст»-2013
Эта работа заняла первое место в номинации «Своя работа» конкурса «био/мол/текст»-2013.
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific. Спонсор приза зрительских симпатий — фирма Helicon.
С увеличением средней продолжительности жизни все больше людей страдают от болезни Альцгеймера и болезни Паркинсона. К сожалению, годы исследований пока не привели ученых к открытию причин развития этих заболеваний и возможной терапии. Это связано, главным образом, с тем, что почти ничего не известно о факторах, вызывающих болезнь, а также с тем, что очень мало пациентов имеют генетическую предрасположенность. Чаще всего эти заболевания являются спорадическими, т.е. причины их возникновения не установлены. Это приводит к бесконечным спорам — никто не знает, как искусственно вызвать это заболевание у модельных животных для экспериментов и поиска лекарств. Поэтому все больше ученых обращают свое внимание на генетические заболевания нервной системы, такие как болезнь Хантингтона (БХ). Это заболевание также, как болезнь Альцгеймера и болезнь Паркинсона, относится к группе нейродегенеративных заболеваний, с которыми его объединяет ряд схожих черт: гибель нейронов центральной нервной системы, накопление амилоидоподобных агрегатов белков, когнитивные и двигательные нарушения у больных. При этом БХ имеет важное преимущество с точки зрения исследователей, т.к. известно, какая мутация вызывает это заболевание. Это дает возможность создавать точные генетические модели и исследовать их на животных. Это важно, потому что если мы поймем патогенез болезни Хантингтона, то нам легче будет разобраться и со спорадическими нейродегенеративными заболеваниями. Это мы и попытались сделать в своем исследовании.
Болезнь Хантингтона
Болезнь Хантингтона (БХ, в русскоязычной литературе также «болезнь Гентингтона») — наследственное заболевание нервной системы, которое поражает примерно 1 из 10 тыс. людей. Болезнь была впервые описана Джорджем Хантингтоном (George Huntington) в 1872, и с тех пор носит его имя, однако клинические симптомы этого заболевания были известны еще в XVI веке под названием «хорея» (от лат. choreus — танец). К признакам хореи относили непроизвольные, нескоординированные быстрые движения, похожие на судороги; именно так описывают и современные медики моторные нарушения, характерные для БХ. Болезнь может порой длиться до двадцати лет, но исход неизменно один и тот же: больной теряет способность самостоятельно передвигаться, говорить, а затем и мыслить. Как правило, симптомы болезни Хантингтона проявляются в возрасте от 30 до 50 лет, хотя у 5–10% пациентов отмечается появление симптомов в возрасте до 20 лет — так называемая ювенильная форма заболевания [1].
Первый симптом болезни Хантингтона — непроизвольные подёргивания конечностей, торса и лицевых мышц. Довольно часто они сопровождаются резкими сменами настроения, депрессией, раздражительностью, неразборчивостью речи и неуклюжестью движений. По мере прогрессирования болезни, к этим симптомам добавляются затруднения или боль при глотании, неустойчивость походки, потеря равновесия, нарушение мыслительных функций и ухудшение памяти. В конце концов, больной теряет способность передвигаться без помощи посторонних и умирает обычно от пневмонии, остановки сердца или других осложнений.
Важной для врачей и исследователей особенностью БХ является то, что это заболевание является наследственным и вызывается мутацией в одном-единственном гене. Оказалось, что к развитию БХ приводит увеличение количества повторов триплета CAG, кодирующего глутамин, в первом экзоне гена белка хантингтина. При этом, чем больше количество повторов этого триплета, тем раньше начинается развитие заболевания. В норме в человеческой популяции встречается от 10 до 35 повторов. У пациентов с БХ количество повторов может быть от 36 до 121, при ювенильной форме — от 50 и выше [2]. Благодаря выявлению генетической основы заболевания, диагностика БХ в настоящее время не представляет проблемы; кроме того, возможной стала пренатальная диагностика заболевания и проверка эмбрионов перед имплантацией при ЭКО, которая позволяет иметь здоровых детей даже носителям мутантного гена.
К сожалению, выявление точной мутации все еще не позволяет ученым определить причину развития болезни Хантингтона и найти соответствующее лечение. Появление в клетке мутантного гена и, соответственно, измененного (мутантного) белка может привести к развитию патологии двумя путями: потеря функции (loss-of-function) или приобретение функции (gain-of-functin). В первом случае мутантный белок не может выполнять ту же функцию, что белок нормальный, и это приводит к нарушению клеточных процессов. Во втором случае, мутантный белок мешает нормальной жизнедеятельности клетки, начиная выполнять какую-то «лишнюю функцию». Чтобы разобраться, что происходит при БХ, ученые интенсивно изучают как функцию нормального белка хантингтина, так и поведение его мутантной формы [3].
К сожалению, попытки определить точную клеточную функцию хантингтина пока не увенчались успехом. Различные исследования указывают на участие этого белка в широком спектре биологических процессов, включая транспорт белков и везикул (мембранных пузырьков-транспортеров), организацию цитоскелета, клатрин-опосредованный эндоцитоз, постсинаптический сигналинг, регуляцию транскрипции и анти-апоптотические процессы [4]. Если удастся доказать, что нарушение какой-либо из этих функций является ключевым для развития заболевания, то лекарственные препараты для поддержания этой функции могут спасти пациентов с болезнью Хантингтона.
Если верна гипотеза о приобретении функции, особое внимание стоит обратить на поведение мутантной формы хантингтина. Оказалось, что мутантный белок формирует агрегаты, которые являются одной из характерных черт развития БХ как у людей,так и у модельных животных (см.врезку). Сначала агрегаты были описаны только в ядре, однако последующие работы выявили их также в цитоплазме и отростках нейронов [5]. В последние годы многие авторы склоняются к тому, что образование агрегатов несет скорее протективную функцию, а основной патогенной формой мутантного хантингтина является мономерный растворимый белок [6].
Модели для изучения болезни Хантингтона
Модели БХ на животных появились более 30 лет назад. Первыми были модели, основанные на введении в стриатум нейротоксических веществ (например, хинолиновой кислоты — агониста NMDA-рецепторов), которые вызывали гибель нейронов. В настоящее время большинство исследователей работает на моделях трансгенных животных, среди которых есть не только мыши и крысы, но и беспозвоночные животные — мушка Drosophila melanogaster и червь Caenorhabditis elegans.
Мышиные модели болезни Хантингтона отличаются друг от друга количеством CAG-повторов и уровнем экспрессии трансгена — искусственно внесенного гена хантингтина. Т.к. именно от этих факторов зависит развитие БХ, разные линии мышей отличаются друг от друга скоростью развития патологий. К наиболее широко используемым моделям относят линии мышей R6/2, R6/1 и YAC128, которые были использованы и в нашей работе. У мышей этих линий симптомы заболевания наиболее выражены и проявляются достаточно быстро. Кроме того, у этих животных с возрастом прогрессируют когнитивные и моторные нарушения, развивается частичная потеря нейронов в стриатуме и коре.
Еще одним из способов моделирования БХ является использование клеточной культуры. В самом простом случае используются культуры клеток со стабильной трансфекцией гена хантингтина. Например, это клетки линии PC12, содержащие индуцибельный трансген первого экзона хантингтина или нейроны стриатума с экспрессией фрагментов хантингтина разной длины. Кроме того, можно использовать первичные культуры из нейронов трансгенных мышей или иммортализованные нейроны.
Почему мы решили исследовать параметры синаптической передачи при болезни Хантингтона
Синаптическая передача — это передача сигналов между нейронами с помощью синаптического контакта. При возбуждении одного нейрона его синаптическое окончание выделяет в синаптическую щель медиатор — химическое вещество, которое оказывает свое возбуждающее или тормозящее воздействие на синаптическое окончание второго нейрона (рис. 1). Таким образом, синапсы связывают нейроны между собой, обеспечивая нормальное функционирование нейронных сетей и всей нервной системы. Если какая-то из систем головного мозга перестает функционировать, причина может крыться либо в нарушении работы отдельных нейронов, либо в нарушении связи между ними, т.е. нарушении синаптической передачи.
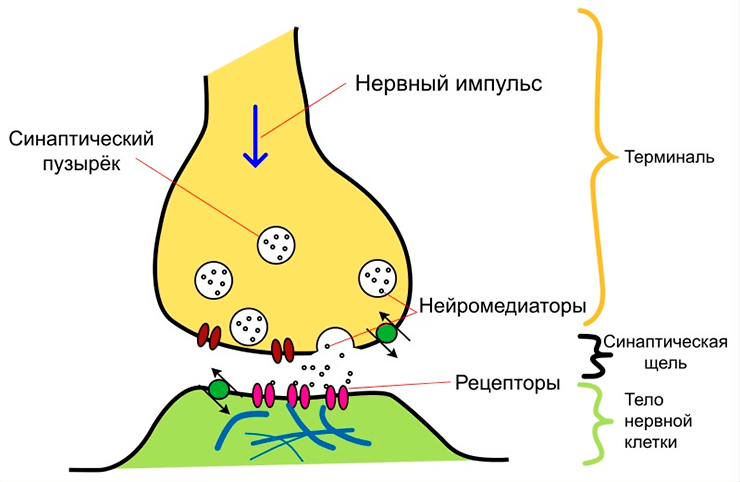
При болезни Хантингона поражается специфическая область головного мозга, называемая стриатумом. Стриатум является частью важного нейронного пути — экстрапирамидной системы, которая участвует в управлении движением и поддержании мышечного тонуса. Гибель нейронов стриатума при болезни Хантингтона приводит к разрушению экстрапирамидной системы, что связано с потерей контроля над движениями у больного человека. Но когда возникают первые патологические симптомы (тремор, нарушение координации), головной мозг человека еще не поврежден: нейроны начинают погибать только через несколько лет после начала развития заболевания. Т.е. болезнь начинается, когда что-то меняется в работе самих нейронов или в синаптической передаче, и эти нарушения впоследствии ведут к гибели нейронов и необратимым последствиям.
Накопившиеся за последние годы результаты исследований заставляют многих ученых склоняться к мысли, что именно нарушение нормальной работы системы нейрональной связи, синапсов и синаптической передачи, приводит к ранним нарушениям в работе экстрапирамидной системы. Оказалось, что в нейронах с мутациями в гене, кодирующем белок хантингтин, наблюдается целый ряд патологических изменений, которые нарушают синаптическую передачу. В таких мутантных клетках нарушается формирование и обновление запаса везикул (пузырьков с медиатором), изменяется внутриклеточная концентрация кальция , который необходим для нормального высвобождения медиатора в синаптическую щель, снижается количество ряда необходимых для функционирования синапса белков и т.п. [7]. Все это приводит к сниженному выбросу медиатора в синаптическую щель, а если медиатора недостаточно, то нейроны начинают хуже «слышать» друг друга, и команды, посылаемые корой головного мозга, не будут выполняться по всей строгости.
В 2013 году Нобелевской премии по физиологии и медицине удостоены работы, благодаря которым стали ясны детали везикулярного транспорта — процесса образования и транспортировки мембранных пузырьков (везикул) между клетками: «Нобелевская премия по физиологии и медицине (2013): везикулярный транспорт» [8]. — Ред.
Изучение нарушенной синаптической передачи при БХ было темой нашего исследования. Может ли быть, что неправильная работа нейронов стриатума на ранней стадии БХ вызвана тем, что они «не слышат» команды нейронов коры? Может ли ослабление синаптической связи приводить к необратимым изменениям в нейронах стриатума и вести к их гибели? О чем мы узнали во время поиска ответов на эти вопросы, рассказано ниже.
Результаты исследований: изменения в синаптической передаче при болезни Хантингтона
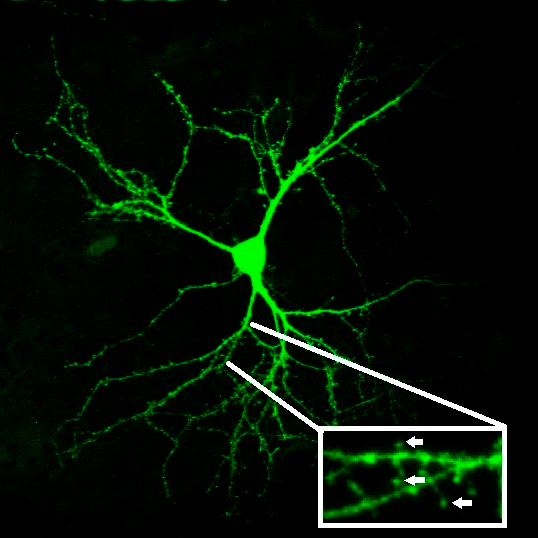
Изучать синаптическую передачу можно различными способами. Например, это можно делать, проникнув в нейронную цепочку с помощью методов электрофизиологии. Нейрон проявляет свою активность с помощью электрического тока, который можно измерить. Если экспериментатор возьмет цепочку из двух нейронов и, активировав один нейрон, зарегистрирует электрическую активность второго, он сможет выяснить, насколько хорошо проходит сигнал. Другой способ изучать функционирование синаптической передачи — исследовать морфологию нейрона. Дело в том, что многие нейроны (в том числе, нейроны коры и стриатума) имеют особые выросты мембраны — шипики, которые нужны им именно для образования синапсов (рис. 2). Чем более активно нейрон «общается» со своими соседями, тем больше на его поверхности шипиков. Взяв на вооружение эти два подхода, мы решили исследовать, как работает синаптическая передача при БХ.
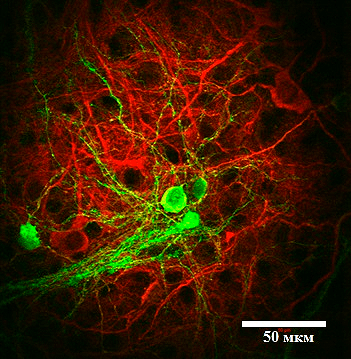
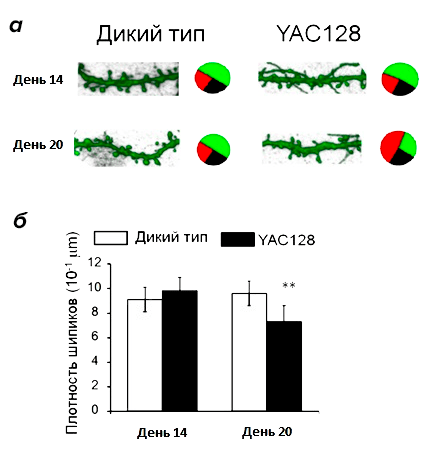
В качестве модели для изучения болезни Хантингтона была использована клеточная культура из нейронов коры и стриатума. Для приготовления культуры незрелые нейроны из изучаемых зон мозга мышей высаживаются в чашки Петри, где они формируют полноценные нейрональные отростки и нейронные цепочки (рис. 3). Использовались мыши дикого типа (без мутаций) и мыши линии YAC128, которые несут мутацию в гене белка хантингтина и являются признанной моделью БХ. На 14–15 день после высаживания нейронов в чашку Петри они достигают зрелого состояния, соответствующего состоянию нейронов в мозге взрослого человека, а на 19–20 день нейроны считаются «старыми»: в них наблюдается ряд клеточных процессов, характерных для мозга пожилых людей. Кроме того, с возрастом в нейронах мышей YAC128 происходит накопление мутантного белка хантингтина и его агрегатов, поэтому изучение нейрональной культуры на этих двух этапах дает представление о том, что происходи в мозге пациента с БХ на ранней и на поздней стадиях заболевания.
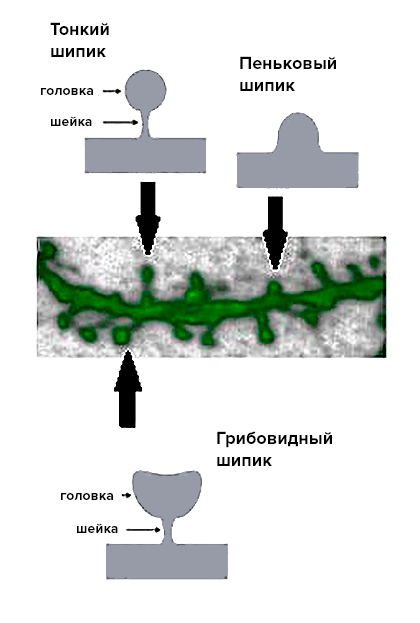
Для начала мы исследовали морфологические отличия между двумя линиями, т.е. сравнили их внешний вид. Для нормальных нейронов стриатума характерно наличие большого количества шипиков, благодаря чему их называют средними шипиковыми нейронами (СШН). Именно шипики формируют большую часть синаптических контактов между нейронами стриатума и коры, и для осуществления нормальной синаптической передачи важно наличие определенного их количества. Так же важно и «качество» шипиков: в современной нейробиологии их разделяют на три группы согласно размеру и форме (рис. 4): грибовидные, тонкие и пеньковые. При этом шипики разных типов выполняют разные функции: считается, что только грибовидные шипики формируют активные синапсы, в то время как тонкие и пеньковые контактов с другими нейронами не образуют. Таким образом, для нормального функционирования нейронной цепочки и эффективной передачи информации по ней необходимо наличие определенного количества грибовидных шипиков.
Для того, чтобы узнать, сколько грибовидных шипиков должно быть на СШН в норме, на всех этапах морфологического анализа как контроль использовалась культура нейронов из головного мозга мышей дикого типа. Оказалось, что количество шипиков на СШН стриатума на 14 день культивирования (молодые нейроны) одинаково в культурах YAC128 и дикого типа, но на 20 день (у «старых» нейронов) наблюдаются значительные изменения (рис. 5). В «постаревшей» культуре YAC128 снижается общее количество шипиков, причем относительное количество грибовидных шипиков, которые образуют активные синапсы, уменьшается в два раза [10]. Получается, что морфологические изменения нейронов, свидетельствующие о нарушении синаптической передачи, развиваются только к «старости» (на поздней стадии заболевания). Значит, на ранних этапах корень проблемы должен лежать в другой области.
Поэтому мы сравнили электрофизиологические характеристики двух линий. Это возможно благодаря электрической активности нейронов, которую можно зарегистрировать с помощью специального метода, называемого пэтч-кламп. Для этого к поверхности нейрона прикладывают тонкую стеклянную пипетку, внутри которой находится электрод. Когда нейрон активируется, на его клеточной мембране изменяется напряжение, и это изменение регистрируется электродом. Если взять два связанных синаптическим контактом нейрона и, возбуждая один, регистрировать ответную электрическую активацию на втором, можно измерить эффективность передачи сигнала через синапс (рис. 6). Если ответная активация возникает не всегда, то, вероятно, синаптическая передача ослаблена. Этот метод может выявить нарушения работы синапса до изменений морфологии нейрона.

Чтобы активировать нейрон, можно использовать несколько способов. Можно добавить в окружающую его жидкость химическое вещество, открывающее ионные каналы, что приводит к электрическому возбуждению нейрона. Можно стимулировать нейрон электрическим током. Но мы для своих экспериментов выбрали более тонкий инструмент воздействия на нейроны, а именно — оптогенетику. Этот подход основан на внесении в нейроны специальных светочувствительных белков — опсинов, в результате чего и сами нейроны становятся чувствительными к свету (рис. 7). В результате, освещая нейроны светом определенной длины волны, можно изменять их активность. Освещение синим светом возбуждает нейрон, а желтым — вызывает торможение (т.е. подавляет активность нейрона).
Оптогенетические технологии в наши дни обещают даже вернуть зрение людям с дегенеративными поражениями сетчатки, придав световую чувствительность не разрушенным фоторецепторам, а клеткам-ганглиям: «Оптогенетика + голография = прозрение?» [11]. — Ред.
Оптогенетика
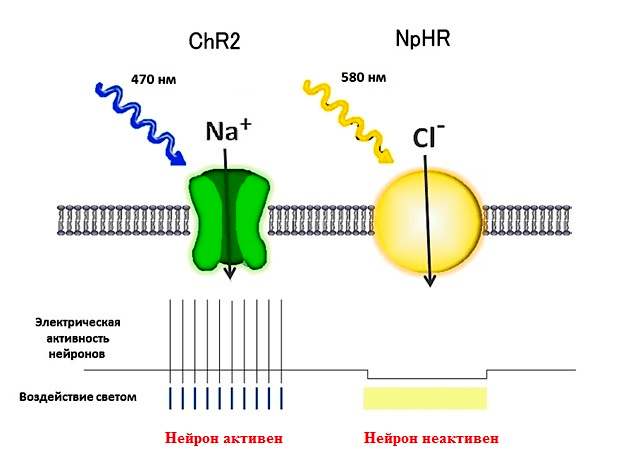
Оптогенетика — метод, объединяющий подходы генетики и оптики для тонкого контроля электрической активности электровозбудимых клеток (нейронов и мышечных волокон) [9]. Для этого в изучаемые клетки вводят гены специальных светочувствительных белков — микробных опсинов, которые являются ионными каналами или насосами (рис. 7). Первая работа, показавшая возможность управлять электрической активностью нейронов при использовании опсина, была опубликована в 2005 году. За последующие несколько лет появился еще ряд экспериментальных работ, позволивших доработать эту методику и доказать ее применимость в различных экспериментальных условиях.
За последние годы было открыто множество различных опсинов, из которых наибольшее применение в оптогенетике нашли галородопсины и каналородопсины. При доставке гена опсина с помощью методов генной инженерии в нейрон, на плазматической мембране появляются светочувствительные каналы, а сама клетка становится светочувствительной. При действии синего света открывается пора каналородопсина (максимум поглощения — 470 нм), который вызывает движение положительно заряженных ионов внутрь клетки, обеспечивая деполяризацию мембраны нейрона и генерацию потенциалов действия. При действии желтого света активируется галородопсин (максимум поглощения — 580 нм), мембрана нейрона гиперполяризуется, вызывая торможение нейрона. Высокое временное разрешение метода оптогенетики позволяет обеспечить очень тонкую регуляцию синаптических событий и является, таким образом, важным инструментом для изучения межнейронных связей.
Совместное применение оптогенетики и классических методов электрофизиологии позволяет извлечь выгоду из положительных качеств каждого из этих подходов. Точность электрофизиологической регистрации объединяется с возможностью использовать световые стимулы разной длительности и интенсивности, что помогает ученым подробно изучать работу нейронных связей.
Электрическая активность нейронов выражается в резких скачках напряжения на клеточной мембране и появлении вследствие этого электрического тока. Эти резкие скачки называются спайками или потенциалами действия и длятся несколько миллисекунд (рис. 8). Мы выяснили, что чем дольше нейрон освещается синим светом, тем больше спайков он за это время создает. Если освещаемый нейрон связан синаптическим контактом с другим нейроном, то на втором нейроне можно зарегистрировать ответную активность — тоже в виде отдельных спайков.
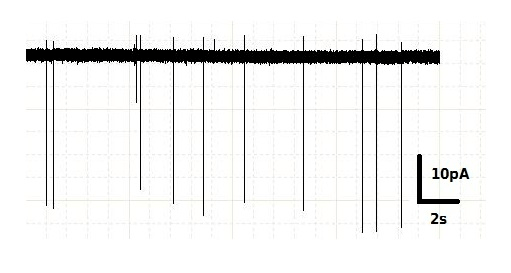
В наших экспериментах мы использовали пару молодых (14 дней) нейронов коры и стриатума, связанных синаптическим контактом. Нейрон коры активировали синим светом, а на нейроне стриатума производили регистрацию ответной активности. Оказалось, что для возникновения ответа на нейроне стриатума нужна определенная длительность освещения (порог активации). Если длительность освещения была ниже порогового значения, то спайк на нейроне стриатума возникал в ответ не на каждую вспышку света. И что самое важное: порог активации для нейронов из мозга здоровых мышей отличался от мышей YAC128 (с мутацией в гене хантингтина). Наиболее ярко эта разница была видна при 50%-активации нейрона стриатума, т.е. длительности освещения, при которой спайк возникает в ответ на каждую вторую вспышку света. Порог 50%-активации нейрона стриатума в ответ на облучение кортикального нейрона для культуры клеток YAC 128 был примерно в два раза (точнее в 2,3±0,8) выше по сравнению с положительным контролем [10].
Получается, что мутация в белке хантингтине приводит к тому, что синаптическая передача ухудшается уже в молодых нейронах, и при этом нарушения происходят на функциональном уровне (без морфологических изменений). Может ли быть так, что именно эти функциональные нарушения в дальнейшем приводят к появлению морфологических изменений, таких как исчезновение шипиков у старых нейронов стриатума?
Чтобы ответить на этот вопрос, мы снова обратились за помощью к оптогенетике, но теперь с ее помощью мы подавляли светом активность нейронов (рис. 9а). Если наше предположение верно, то временное отсутствие активирующего воздействия коры должно привести к исчезновению шипиков на нейронах стриатума в культуре YAC128. Действительно, после эксперимента количество шипиков в положительном контроле осталось неизменным, но в культуре YAC128 значительно снизилось (рис. 9б, в). Получается, что моделирующие БХ нейроны особенно чувствительны к ослаблению активирующего воздействия нейронов коры, поэтому длительное ослабление синаптической связи между этими нейронами приводит к уменьшению количества шипиков на 20 день культивирования [10].
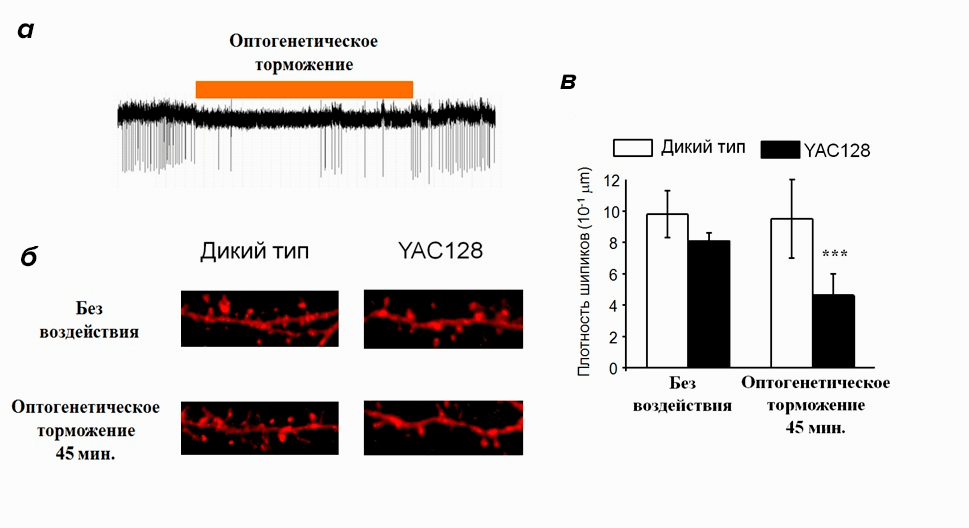
Таким образом, мы обнаружили, что в нашей модели БХ наблюдается нарушение синаптической передачи, которое развивается в два этапа. На ранней стадии (молодые нейроны, на 14 день культивирования нейронов) происходит функциональное ослабление сипаптической связи, которое на более поздней стадии (старые нейроны, на 20 день культивирования) приводит к морфологическим нарушениям синаптических контактов.
Вывод: нарушение синаптической передачи и нейродегенеративные заболевания
Исследование подтвердило нашу первоначальную догадку: нарушение работы стриатума при БХ связано, прежде всего, с нарушением синаптической передачи. Нейроны стриатума перестают «слышать» команды нейронов коры, и, в результате, человек начинает терять контроль над своими движениями. Но на ранней стадии заболевания эти нарушения носят функциональный характер и, вероятно, обратимы. Если выяснить причины, ведущие к нарушению работы синапсов, то возможно разработать лекарственное средство, которое поможет остановить патологические изменения, обеспечит нейронам стриатума необходимый уровень активации и, таким образом, предотвратит развитие необратимых морфологических нарушений.
Результаты этой работы подталкивают и к другому важному заключению: при изучении нейродегенеративных заболеваний ученым следует обратить больше внимания на синаптическую передачу и взаимодействие нейронов. Мы знаем достаточно много о том, как влияет накопление амилоидных агрегатов при болезни Альцгеймера на жизнедеятельность клетки, и какие нейроны погибают при развитии болезни Паркинсона, но это все еще не привело к появлению эффективной терапии. Возможная причина этих неудач — мы не учитываем последствия нарушения синаптических связей — те, которые мы открыли при исследовании болезни Хантингтона. Хочется надеяться, что наши находки подскажут исследователям направление для разработки эффективных способов лечения болезни Хантингтона, и Тринадцатая наконец-то будет здоровой!
Исследование было проведено в лаборатории молекулярной нейродегенерации СПбГПУ (зав. лаб. — проф. Техасского университета Илья Борисович Безпрозванный) под руководством к.б.н. Дмитрия Николаевича Артамонова.
Литература
- Hayden M.R. Huntington’s Chorea. New York: Springer, 1982;
- DR Langbehn, RR Brinkman, D Falush, JS Paulsen, MR Hayden, on behalf of an International Huntington’s Disease Collaborative Group. (2004). A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clinical Genetics. 65, 267-277;
- Идентифицированы белки, «слипающиеся» при болезни Гентингтона;
- Sara Imarisio, Jenny Carmichael, Viktor Korolchuk, Chien-Wen Chen, Shinji Saiki, et. al.. (2008). Huntington’s disease: from pathology and genetics to potential therapies. Biochem. J.. 412, 191-209;
- Claire-Anne Gutekunst, Shi-Hua Li, Hong Yi, James S. Mulroy, Stefan Kuemmerle, et. al.. (1999). Nuclear and Neuropil Aggregates in Huntington’s Disease: Relationship to Neuropathology. J. Neurosci.. 19, 2522-2534;
- Boxun Lu, James Palacino. (2013). A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. The FASEB Journal. 27, 1820-1829;
- Benjamin Ray Miller, Ilya Bezprozvanny. (2010). Corticostriatal circuit dysfunction in Huntington’s disease: intersection of glutamate, dopamine and calcium. Future Neurology. 5, 735-756;
- Нобелевская премия по физиологии и медицине (2013): везикулярный транспорт;
- Karl Deisseroth. (2011). Optogenetics. Nat Methods. 8, 26-29;
- Артамонов Д.Н., Коржова В.В., Ву Д., Рыбальченко П.Д., Им К., Красноборова В.А., Власова О.Л., Безпрозванный И.Б. (2013). Нарушения синаптической передачи при болезни Хантингтона на кортикостриатной модели культуры нейронов. «Биологические мембраны». 30, 1–13;
- Оптогенетика + голография = прозрение?.
Источник https://akonit-med.ru/articles/nevrologiya/17911-prichiny-simptomy-i-lechenie-patologii-gentingtona-sklonnost.html
Источник https://chastnaya-psihiatricheskaya-klinika.ru/horeya-gentingtona-lechenie
Источник https://biomolecula.ru/articles/kak-spasti-trinadtsatuiu-perspektivy-lecheniia-bolezni-khantingtona