Фенилкетонурия — причины, симптомы, диагностика и лечение
Фенилкетонурия (фенилпировиноградная олигофрения, ФКУ) — это генетически детерминированное врожденное заболевание, которое приводит к накоплению в организме избытка аминокислоты фенилаланина. Именно по этой причине возникают токсические симптомы заболевания.
Фенилкетонурия в истории медицины впервые описана в 1934 году норвежским врачом Фоллингом. Он осмотрел, в частности, двух братьев и сестер и заметил их аномальные нарушения обмена веществ и симптомы. Исследования показали наличие в детской моче вещества, которое Фоллинг идентифицировал как фенилкетон. Через год это расстройство было описано еще более подробно последующими исследователями и названо фенилкетонурией.
В настоящее время фенилкетонурия относительно хорошо изучена, хотя некоторое время она была основным летальным фактором для детей и вызывала у больных интеллектуальные нарушения.
В настоящее время, благодаря развитию медицины и многочисленным исследованиям заболевания, достаточно хорошо известны патогенез фенилкетонурии, ее лечение, профилактика патологических последствий и изменений в организме в результате заболевания.
Причины болезни
Средняя частота заболеваемости составляет примерно 1 случай на 15 000 новорожденных. Однако есть страны, в которых заболевание встречается чаще, чем в других, например, в Ирландии, где заболевание регистрируется у 1 из 4500 новорожденных.
Фенилкетонурия является одним из генетических заболеваний. Таким образом, это врожденное заболевание, первые последствия которого диагностируются с первых дней после рождения ребенка. Фенилкетонурия вызывается мутацией гена на хромосоме 12. Этот ген отвечает за кодирование фермента фенилаланингидроксилазы. Мутация этого гена в организме человека вызывает нарушение ферментативной активности и работы ранее упомянутого фермента гидроксилазы, отвечающего за превращение аминокислоты фенилаланина в другую аминокислоту — тирозин.
В результате действия такого фактора в крови больного человека начинают накапливаться фенилаланин и отдельные продукты его клеточного метаболизма, при этом возникает достаточно большой дефицит тирозина. В результате таких концентраций отдельных веществ у детей с фенилкетонурией возникают поражения головного мозга и необратимые изменения, которые могут привести, в том числе, к интеллектуальным нарушениям.
Фенилкетонурия — симптомы
Состояние новорожденного ребенка без сразу выявленного заболевания поначалу не вызывает серьезных опасений. У большинства детей с фенилкетонурией светлые волосы и кожа, а также голубые глаза, но это не факторы, прямо указывающие на диагноз, поскольку они являются переменными, обычно связанными с внешним видом.
Большинство симптомов заболевания появляются постепенно и обостряются на 2-3 месяце жизни ребенка. В это время появляются такие симптомы, как:

- торможение психомоторного развития;
- низкая двигательная активность или наоборот — гиперактивность;
- кожная сыпь;
- припадки;
- также может появиться рвота;
- повышенный или пониженный мышечный тонус;
- малыш и, прежде всего, его выделения начинают характерно пахнуть — так называемый «мышиный запах».
Диагностика и лечение
Новорожденных обследуют на фенилкетонурию — берут кровь и назначают анализы. В случае положительного результата, который может указывать на фенилкетонурию, проводится расширенная диагностика. Окончательный диагноз заболевания должен быть поставлен как можно раньше в связи с необходимостью проведения лечения.
Фенилкетонурия является генетическим заболеванием, и для него не существует лекарства или фармакологического лечения. Однако медицина знает способ затормозить развитие болезни и тем самым избежать ее последствий и купировать симптомы. Профилактика поражения головного мозга у детей с фенилкетонурией основана на строгой диете, исключающей фенилаланин и любые продукты, содержащие эту аминокислоту.
Это единственный способ лечения фенилкетонурии.
В дополнение к диете детям необходимо регулярно контролировать уровень фенилаланина в крови. Врачи рекомендуют проводить эти проверки очень часто. Это означает, что у маленьких детей их делают даже раз в неделю или раз в две недели, а после 7 лет — каждый месяц. Однако это во многом зависит от течения болезни и индивидуального случая ребенка.
Наследование болезни
Ребенок может заболеть, если оба родителя больны ФКУ или оба являются носителями. Вероятность заболевания в этом случае оценивается в 25%. Если у одного из родителей есть заболевание или дефектный ген, ответственный за эту мутацию, ребенок обязательно будет здоров.
Сама по себе фенилкетонурия не является противопоказанием к рождению детей. Это связано с тем, что в настоящее время заболевание диагностируется относительно легко, эффективно (особенно в развитых странах Европы) и существуют методы купирования симптомов. Все это влияет на то, что сознательные носители болезни все же решают завести потомство.

Диета для людей с фенилкетонурией
Рацион ребенка, страдающего этим заболеванием, должен разрабатываться совместно с врачом-диетологом и модифицироваться в зависимости от возраста и состояния здоровья больного. Врач определяет, какое количество фенилаланина пациент может безопасно принимать, а также определяет любые дополнительные питательные вещества, которые пациент должен использовать в качестве пищевых добавок (содержащих аминокислоты, витамины и минералы, которые не могут поступать в организм из-за ограниченной диеты).
Продукты, потребляемые детьми с ФКУ, должны содержать лишь небольшое количество фенилаланина. Поэтому из рациона следует исключить:
- любые мясные продукты,
- рыбу,
- яйца,
- сыр,
- молоко и все продукты из него,
- хлеб и другие мучные изделия.
Диета кажется очень ограничительной, и это на самом деле так. Она требует большой систематичности и внимания со стороны всей семьи, но это единственный способ лечения болезни. Тот факт, что в настоящее время большинство пищевых продуктов имеют информацию о содержании или отсутствии в составе аминокислоты фенилаланина, упрощает дело.
Предполагается, что диета должна полностью соблюдаться примерно до 18-20 лет. Это период наиболее эффективного развития головного мозга — в последующие годы жизни с согласия врача и контроля параметров, связанных с заболеванием, можно попробовать расширить рацион небольшими количествами фенилаланина в продуктах.
Эта запись была размещена в Справочник заболеваний,Ф. Добавить в закладки постоянная ссылка.
Все представленные на сайте материалы предназначены исключительно для образовательных целей и не предназначены для медицинских консультаций, диагностики или лечения. Администрация сайта, редакторы и авторы статей не несут ответственности за любые последствия и убытки, которые могут возникнуть при использовании материалов сайта.
Фенилкетонурия

Фенилкетонурия представляет собой генетическую патологию, которая характеризуется нарушением аминокислотного обмена, накоплением фенилаланина и продуктов его метаболизма в тканях и биологических жидкостях, приводящим к тяжелым поражениям центральной нервной системы.
Причины фенилкетонурии
Данное заболевание наследуется по аутосомно-доминантному типу. Для развития патологического процесса достаточно одной полиморфной копии от обоих родителей-гетерозиготных носителей дефектного гена, который кодирует энзим, располагающийся на локусе 12q22-q24.1 длинного плеча 12-ой хромосомы – фенилаланин-4-гидроксилазу.
Патогенез
Наследственный дефицит энзима, который принимает участие в конверсии незаменимой ароматической аминокислоты фенилаланина, происходящей в митохондриях клеток печени, в тирозин – аминокислоту, необходимую для синтеза белков и биологически активных веществ. Ее производными являются:
- тирамин, который клетки организма используют для выработки адреналина, дофамина и норадреналина;
- дийодтирозин, требующийся для производства гормона щитовидной железы – тироксина.
Недостаток фенилалаиин-4-гидроксилазы провоцирует нарушение окисления поступающего с пищей фенилаланина и значительное увеличение его количества в ликворе и крови. Это явление сопровождается разрушением миелиновой оболочки нервных волокон и снижением синтеза нейро-медиаторов, что приводит к органическому поражению головного мозга.
Симптомы
Клинические проявления фенилкетонурии заметны уже спустя 3-5 месяцев с момента рождения ребенка – у него наблюдается:
- вялость;
- частые срыгивания;
- гипер-возбудимость;
- мышечная дистония;
- судорожный синдром.
Прогрессирование патологии сопровождается упорной рвотой, заметной задержкой развития интеллектуальных, двигательных и речевых навыков. Малыш безучастный, не узнает близких, не сидит и не становится на ножки, у него отмечается:
- шелушение кожных покровов;
- исходящий от тела запах сырости;
- позднее появление молочных зубов;
- склеродермия;
- микроцефалия;
- диспластическое телосложение;
- снижение кровяного давления;
- дрожание рук;
- гиперкинезы;
- расстройства пищеварения;
- умственная отсталость.
Методы диагностики
Для дифференциального диагностирования фенилкетонурии разработана специальная программа обследования новорожденных младенцев, включающая:

1. Скрининг-тест, который осуществляют на 3-й день жизни ребенка – образец капиллярной крови отбирают на специальный бланк. При наличии гипер-фенилаланемии (свыше 2,19 мг% аминокислоты) требуется повторное тестирование и консультация генетика.
2. Биохимический анализ крови – для измерения концентрации:
- фенилаланина;
- АСТ;
- тирозина;
- АЛТ;
- катехоламинов.
3. Биохимическое исследование мочи, позволяющее обнаружить кетоновые тела и продукты распада гормонов.
4. Консультацию детского невропатолога.
5. Электроэнцефалографию и магниторезонансную томографию головного мозга.
6. Молекулярно-биологический анализ крови – для обнаружения мутантных вариантов генов.
Лечение
Пациентам с фенилкетонурией необходимо соблюдать диетическое питание, ограничивающее употребление белковой пищи, и принимать:
- поливитамины;
- антиконвульсанты;
- ноотропы;
- гепато-протекторы.
Комплекс лечебных мероприятий включает лечебную физкультуру, массаж, иглорефлексотерапию.
Фенилкетонурия
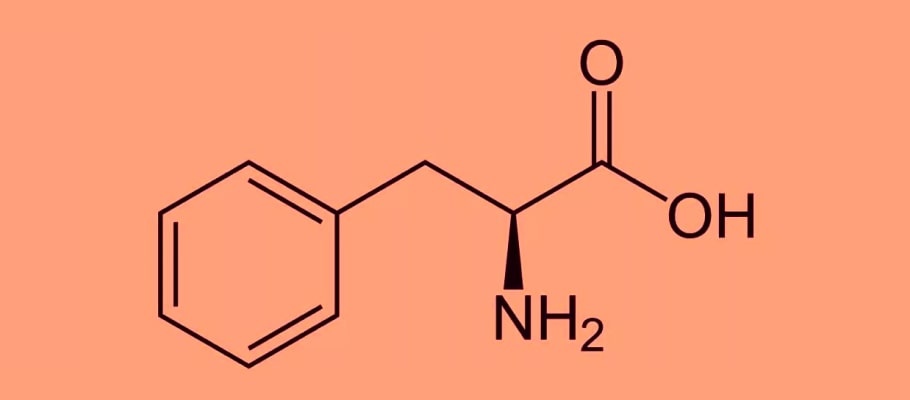
Это наследственное заболевание также называют болезнью Феллинга и относят к группе ферментопатий, обусловленных нарушением метаболизма аминокислот, преимущественно фенилаланина. В случае несоблюдения низкобелковой диеты больные фенилкетонурией могут страдать от тяжелых поражений ЦНС вследствие накопления в организме фенилаланина и его токсических метаболитов.
Пренатальная диагностика моногенного аутосомно-рецессивного заболевания (пробанд — гомозигота, генотип пробанда и родителей известен)
21 рабочиx дней
Проведение пренатальной диагностики позволяет до рождения ребенка обнаружить мутацию и сделать верный прогноз о его здоровье в отношении заболевания.
Причины заболевания

Фенилкетонурия — генетическая патология с аутосомно-рецессивным характером наследования. Чаще всего болезнь Феллинга возникает при мутации гена, который кодирует фермент фенилаланин-4-гидроксилазу и расположен на длинном плече хромосомы 12 (локус12q22-q24). Эта мутация является причиной фенилкетонурии I типа, которая встречается в 98 % всех случаев.
Существуют также атипичные формы заболевания. Это фенилкетонурия II и III типов. При схожей симптоматике они не поддаются коррекции посредством диетотерапии.
Патогенез фенилкетонурии
Дефицит фермента фенилалаиин-4-гидроксилазы лежит в основе классической формы заболевания, он приводит к нарушению окисления поступающего с пищей фенилаланина. Его концентрация в крови и спинномозговой жидкости значительно возрастает, а уровень тирозина падает. Это приводит к нарушению миелинизации нервных волокон, снижению образования нейромедиаторов и запускает патогенетические механизмы задержки умственного развития и вызывает прогредиентное слабоумие.
Симптоматика фенилкетонурии
Манифестация болезни Феллинга происходит в возрасте 2–6 мес. Развиваются первые неспецифические симптомы: вялость, беспокойство и гипервозбудимость, мышечная дистония, срыгивание, судорожный синдром. Патогномоничным симптомом фенилкетонурии считается упорная рвота.
После 6 мес. наблюдается отставание ребенка в психомоторном развитии. Это проявляется как снижение активности, безучастность к окружающему, ребенок перестает узнавать родных, не делает попыток сесть или встать на ноги. Могут появляться шелушение кожи, экзема, дерматит, склеродермия.
Дети с фенилкетонурией, не получающие лечения, страдают от микроцефалии, прогнатии, позднего прорезывания зубов, гипоплазии эмали. Для них характерна задержка речевого развития, к трем–четырем годам развивается олигофрения с практически полным отсутствием речи.
Больные имеют диспластическое телосложение, отличаются светлой кожей, волосами и глазами. Верхние и нижние конечности согнуты в суставах (так называемая «поза портного»), походка шаткая, семенящая, может быть тремор рук, гиперкинезы.
НИПТ расширенная панель
8 рабочиx дней
Неинвазивный пренатальный скрининг хромосомных аномалий плода (НИПТ) на сегодняшний день является рутинной практикой врачей-генетиков и акушеров гинекологов. Он позволяет избежать напрасных волнений, связанных с ложными результатами биохимического скрининга, а также необоснованного проведения инвазивных процедур.
Диагностика фенилкетонурии
Исследование на наличие болезни Феллинга входит в программу неонатального скрининга, который обязателен для всех новорожденных. Тест проводят на 3–5 днях жизни ребенка. При обнаружении гиперфенилаланинемии дают направление на генетическое исследование.
Для подтверждения наличия фенилкетонурии проводят:
- определение концентрации фенилаланина и тирозина в крови;
- анализ активности печеночных ферментов;
- биохимическое исследование мочи;
- МРТ головного мозга;
- ЭЭГ;
- обследование у детского невролога.
Генетический дефект может быть обнаружен еще до рождения в рамках неинвазивного пренатального ДНК-теста Panorama или неинвазивного пренатального ДНК-скрининга, которые можно пройти в медико-генетическом центре «Геномед». После рождения ребенка можно провести поиск мутаций в гене РАН (в том числе расширенный), сделать «Скрининг на наследственные заболевания», панель «Наследственные нарушения обмена веществ».
Лечение, прогноз и профилактика фенилкетонурии
Основа терапии — соблюдение диеты с низким содержанием белков. Комплексное лечение может включать в себя прием витаминно-минеральных комплексов, ноотропных средств, антиконвульсантов, а также ЛФК, массаж, иглорефлексотерапию.
При раннем обнаружении фенилкетонурии и назначении элиминационной диеты прогноз благоприятный. Поздно начатая терапия имеет неблагоприятный прогноз по отношению к умственному развитию.
Профилактика фенилкетонурии включает в себя пренатальное генетическое исследование. Женщины с болезнью Феллинга должны тщательно соблюдать диету до начала и во время беременности.
Источник https://el-klinika.ru/fenilketonuriya-prichiny-simptomy-diagnostika-i-lechenie/
Источник https://medcentr-endomedlab.ru/zabolevanija/fenilketonuriya.html
Источник https://genomed.ru/journal/geneticheskie-zabolevaniya/fenilketonuriya