Что такое мукополисахаридоз?
Мукополисахаридозы (МПС) – группа редких наследственных заболеваний обмена веществ, приводящих к прогрессирующему поражению множества органов и тканей. Всего выделяют 6 типов мукополисахаридозов, каждый из которых характеризуется проблемой, вызванной определенным ферментом. Данный ресурс посвящен мукополисахаридозу I типа, связанному с недостаточностью фермента альфа-L-идуронидазы.
Еще в утробе матери нехватка фермента начинает приводить к нарушению нормального развития органов и тканей, которое прогрессируют по мере роста ребенка после рождения. Деформация скелета, патология суставов, клапанов сердца, частые респираторные заболевания, поражение нервной системы вплоть до умственной отсталости – вот неполный перечень проявлений мукополисахаридоза I типа. В итоге ребенок может стать инвалидом. 1
Патологические изменения, лежащие в основе заболевания.
Мукополисахаридозы представляют собой группу из 11 редких наследственных (т.е. передающихся от родителей к детям) заболеваний, которые связаны с нарушением обмена гликозаминогликанов. Данные вещества присутствуют в организме человека повсеместно и заполняют пространство между клетками, поэтому нарушение их метаболизма приводит к серьезным нарушениям функций практически каждого органа. В зависимости от гена, который поражается при мукополисахаридозе, определяется тип заболевания. Всего выделяют 11 генов, поломки (мутации) в которых приводят к снижению или потере функции определенных ферментов, разрушающих гликозаминогликаны и приводящих к развитию мукополисахаридоза какого-либо типа. В настоящее время только 4 из 11 типов мукополисахаридозов поддаются лечению: мукополисахаридоз I, II, IV и VI типов. Своевременно начатая терапия позволяет улучшить качество жизни заболевшего и предотвратить серьезные нарушения функций организма.
Мукополисахаридоз I типа (МПС I) – как и другие типы мукополисахаридозов, является наследственным заболеванием, причиной которого является унаследованный от обоих родителей (в большинстве случаев, здоровых) поломанные гены. Из-за генных изменений возникает дефицит фермента (альфа-L-идуронидаза), приводящий к накоплению мукополисахаридов (или гликозаминогликанов) в соединительной ткани всех органов – в центральной нервной системе, сердце, сосудах, опорно-двигательном аппарате, печени, селезенке, органах зрения и слуха.

В зависимости от проявлений заболевания выделяют разные по степени тяжести формы МПС I (см. Признаки и симптомы мукополисахаридоза) 10
Клиническая картина заболевания
Поскольку гликозаминогликаны накапливаются практически во всех органах и тканях, заболевание имеет множество клинических появлений. Наиболее часто родители обращаются к врачу в связи с одним или несколькими проявлениями мукополисахаридоза у ребенка:
- суставы: тугоподвижность суставов рук, изменение их формы, напоминающее ревматоидный артрит без признаков воспаления; 6, 7
- сердце: прогрессирующая недостаточность клапанов сердца, чаще всего митрального с потерей его функции; 8
- нервная система: отек оболочки «сдавливает» головной мозг, приводя к повышению внутричерепного давления. Также поражаются клетки самого мозга, что в итоге проявляется в виде прогрессирующего поражения нервной системы, вплоть до умственной отсталости; 1
- органы дыхания: откладывающиеся гликозаминогликаны увеличивают «размеры трахеи», сужая ее просвет, и являются причиной утолщения голосовых связок. В результате нормальная циркуляция воздуха затрудняется, что приводит к развитию частых респираторных заболеваний у таких детей; 1
- внешние проявления: изменение черт лица (грубые, крупные черты лица, выступающий подбородок, большой рот), короткая утолщенная шея, большой язык, оставляющий рот приоткрытым. 9
Симптомы могут различаться в зависимости от формы заболевания.
Центры экспертизы
Надеемся, вы смогли найти нужную информацию, но для постановки точного диагноза рекомендуем обратиться к специалисту.

Показать источники
- 1. Мукополисахаридоз I типа у детей. Клинические рекомендации. Союз педиатров России, 2016. Доступ: pediatr-russia.ru от 26.02.2019.
- 2. Neufeld EF & Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. 2001. McGraw Hill, New York, USA.
- 3. Clarke LA. Expert Rev Mol Med 2008; 10: e1.
- 4. Neufeld EF & Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. 2001. McGraw Hill, New York, USA.
- 5. Simmons MA, et al. Int J Pediatr Otolarygol 2005; 69: 589–595.
- 6. Cimaz R, et al. Attenuated type I mucopolysaccharidosis in the differential diagnosis of juvenile idiopathic arthritis: a series of 13 patients with Scheie syndrome. Clin Exp Rheumatol. 2006 Mar-Apr;24(2):196-202.
- 7. Rolando Cimaz, et al. Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis; Pediatric Rheumatology 2009, 7:18.
- 8. Maria Veronica Munoz-Rojas, Nicolas Pangaud, Vassili Valayannopoulos. Awareness of MPS I among cardiologists. Molecular Genetics and Genomics. February 2018Volume 123, Issue 2, Page S101.
- 9. Clarke LA. Mucopolysaccharidosis Type I. GeneReviews. February 2016. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1162/. Accessed April 2018.
- 10. Stéphane Mitrovic, Hélène Gouze, Laure Gossec, Thierry Schaeverbeke, Bruno Fautrel, Mucopolysaccharidoses seen in adults in rheumatology; Joint Bone Spine 84 (2017) 663-670.
- Официальное уведомление
- Обратная связь
- Карта сайта
- Для специалистов здравоохранения
- Санофи
© АО «Санофи Россия».
125009, Москва, ул. Тверская, д. 22. Телефон: +7 495 721 14 00
Дата последнего обновления: 23.03.2023 «Все права защищены»
*Под диагностикой и лечением понимаются исключительно диагностика и лечение специалистом. Пожалуйста, проконсультируйтесь с Вашим лечащим врачом.
Дифференциальная диагностика мукополисахаридозов
О мукополисахаридозах (МПС), редких наследственных заболеваниях обмена веществ из группы лизосомных болезней накопления, в последние годы говорят часто. Еще недавно схожесть симптоматики МПС с другими заболеваниями и недостаточная лабораторная диагностика часто приводили к ошибочным диагнозам и неадекватному лечению. Вместе с тем ранняя диагностика мукополисахаридозов и правильная терапия во многом определяют прогноз заболевания и перспективы пациента.
Основа патогенеза МПС — накопление в органах и тканях патологического субстрата, а именно гликозаминогликанов (ГАГ), или мукополисахаридов, которые являются базовым компонентом соединительной ткани. Избыточное накопление ГАГ приводит к дисфункции клеток, органов и тканей и вызывает разнообразные клинические проявления. МПС относится к мультисистемным заболеваниям, поэтому дифференциальная диагностика данной нозологии актуальна для врачей любых специальностей, особенно педиатрического профиля.

Опытом правильной диагностики МПС (на примере синдрома Хантера как наиболее распространенного типа мукополисахаридоза) поделилась старший научный сотрудник лаборатории редких наследственных болезней у детей Центра редких болезней ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России, кандидат мед. наук Татьяна Подклетнова.
Объективная лабораторная диагностика
Основа диагностики — клинические проявления заболевания. Все типы МПС имеют общие внешние клинические признаки, которые могут и должны вызвать у врача на амбулаторном приеме настороженность в отношении МПС: гаргоилизм, костные изменения, органомегалия и т. д.
Лабораторная диагностика включает три этапа.
1. Количественная и качественная оценка экскреции ГАГ в моче. Неспецифический тест, который с высокой степенью вероятности позволяет заподозрить у пациента МПС, не дает, однако, четкой информации о типе заболевания.
При легких формах заболевания избыточное количество ГАГ в моче может не определяться, следовательно, тест будет ложно-отрицательным. На основании только этого лабораторного анализа ставить окончательный диагноз нельзя!
2. Энзимодиагностика. Измерение активности фермента в лейкоцитах крови. При МПС любого типа снижается уровень определенного фермента в лейкоцитах или плазме крови. В частности, при МПС 2-го типа снижается уровень идуронат-2-сульфатазы в плазме (норма: 250–620 nM/mg/h); МПС 2-го типа: 0–20 nM/mg/h); при МПС 1-го типа — α-L-идуронидазы в периферической крови (норма: 61–176 nM/mg/h; МПС 1-го типа: 0–18 nM/mg/h) и т. д. Однако существует ряд заболеваний, при которых уровень ферментов в крови также может снижаться, например, множественная сульфатазная недостаточность. Необходим третий этап.
3. Молекулярно-генетическая диагностика. Наличие мутаций в том или ином гене дает окончательный ответ и возможность установить объективный диагноз. Так, МПС 1-го типа обусловлен мутацией в гене IDUA, МПС 2-го типа — мутацией в гене IDS и т. д.
Существуют и другие методы диагностики, которые обязательно должны быть предложены семьям, планирующим рождение ребенка, но у которых уже есть случаи МПС среди близких родственников. Речь о пренатальной и предимплантационной диагностике.
Пренатальная диагностика проводится между 10-й и 12-й неделями уже наступившей беременности. Выполняется определение ферментной активности в ворсинах хориона, проводится молекулярно-генетическая диагностика плода. Надежность метода достигает 99 %. У родителей есть возможность при выявлении патологии прервать беременность на малом сроке. Или же принять осознанное решение о рождении ребенка с генетическим заболеванием. В этом случае ранняя пренатальная диагностика дает возможность начать адекватное лечение на самом раннем этапе, что, естественно, влечет за собой совершенно иные, более благоприятные прогнозы относительно качества и продолжительности жизни ребенка.
Предимплантационная диагностика проводится перед искусственным оплодотворением и позволяет избежать рождения ребенка с данной генетической патологией.
Шаг 1. Дифдиагностика с другими видами мукополисахаридозов
В современной классификации существуют 7 основных типов мукополисахаридозов. Наиболее часто (помимо МПС 2-го типа) встречаются МПС 1-го, 3-го, 4-го, 6-го типов.
Особенности МПС 1-го типа (синдром Гурлера, синдром Гурлера — Шейе, синдром Шейе):
- гипертрофия десен, редкие мелкие зубы, макроглоссия;
- частые респираторные инфекции, отиты, бронхиты;
- тугоухость;
- помутнение роговицы, пигментная дегенерация сетчатки, глаукома;
- низкий рост, кифосколиоз, деформация грудной клетки, врожденный вывих бедра, контрактуры крупных и мелких суставов;
- синдром запястного канала;
- цервикальный стеноз;
- шумное носовое дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- задержка психомоторного развития, регресс навыков;
- экскреция гепарансульфата и дерматансульфата с мочой.
МПС 1-го типа имеет аутосомно-рецессивный тип наследования, т. е. заболевание одинаково свойственно лицам мужского и женского пола, в то время как МПС 2-го типа — мужское заболевание, девочки болеют крайне редко. Помутнение роговицы при МПС 2-го типа практически не встречается либо очень «нежное», визуально не видное. При МПС-1 помутнение роговицы видно визуально. Костные деформации и проявления при МПС-1 превалируют, ярче выражены, быстрее прогрессируют.
Особенности МПС 3-го типа (синдром Санфилиппо):
- болеют лица мужского и женского пола;
- ведущим является поражение ЦНС: интеллектуальный дефицит, нарушения поведения и сна, двигательные нарушения, эпилепсия;
- высокий рост, кифосколиоз, деформация грудной клетки, контрактуры крупных и мелких суставов менее выраженные;
- частые респираторные инфекции, отиты, бронхиты;
- тугоухость;
- шумное дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- экскреция гепарансульфата с мочой.
Наиболее яркое отличие МПС 3-го типа — более выраженная неврологическая симптоматика по сравнению с другими типами МПС, для которых характерен интеллектуальный дефицит.
Например, при МПС-1 и МПС-2 иногда бывает и так, что ребенок изначально развивается в соответствии с возрастом, лишь затем появляются симптомы задержки и происходит регресс. В случае МПС-3 ребенок с раннего детства имеет прогрессирующую задержку развития, нарушения поведения, к которым позже присоединяются другие неврологические проявления: двигательные нарушения, судороги. Что касается соматических проявлений, то и здесь есть отличия: дети с МПС 3-го типа имеют достаточно высокий рост (другим типам свойственна низкорослость); при этом типе мукополисахаридоза не бывает цервикального стеноза и синдрома запястного канала.
Особенности МПС 4-го типа (синдром Моркио):
- изменения черт лица по типу гаргоилизма отсутствуют;
- частые респираторные инфекции, отиты, бронхиты;
- сохранный интеллект;
- тугоухость;
- помутнение роговицы, пигментная дегенерация сетчатки;
- низкий рост, кифосколиоз, килевидная деформация грудной клетки, грубые деформации крупных и мелких суставов при отсутствии контрактур;
- цервикальный стеноз;
- шумное дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- экскреция кератансульфата с мочой.
Примечательно, что детям с МПС 4-го типа не присущ гаргоилизм. Кроме того, особенностью этого типа МПС являются специфические костные деформации: килевидная деформация грудной клетки, грубая деформация позвоночника, вальгусная деформация конечностей. МПС-4 отличает и абсолютно сохранный интеллект пациента (в отличие от МПС-1 или МПС-2, при которых есть пациенты как с высоким уровнем интеллекта, так и с достаточной грубой задержкой интеллектуального развития).
При ранней постановке диагноза и правильном ведении болезни пациенты с синдромом Моркио могут быть успешно адаптированы в обществе.
Особенности МПС 6-го типа(синдром Марото — Лами):
- гипертрофия десен, редкие, мелкие зубы, макроглоссия;
- частые респираторные инфекции, отиты, бронхиты;
- сохранный интеллект;
- тугоухость;
- помутнение роговицы, пигментная дегенерация сетчатки;
- низкий рост, кифосколиоз, деформация грудной клетки, врожденный вывих бедра, контрактуры крупных и мелких суставов;
- синдром запястного канала;
- цервикальный стеноз;
- шумное дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- экскреция дерматансульфата с мочой.
Этот тип МПС имеет всю симптоматику, характерную для других типов мукополисахаридоза. Из-за видимого помутнения роговицы, грубых костных деформаций, выраженных контрактур суставов МПС 6-го типа похож на тяжелые формы МПС-1 и МПС-4.
Важно отметить, что при данном типе МПС, как и при МПС-4, пациенты имеют сохранный интеллект и при ранней диагностике заболевания, своевременном начале ферментозаместительной терапии могут быть социализированы в обществе.
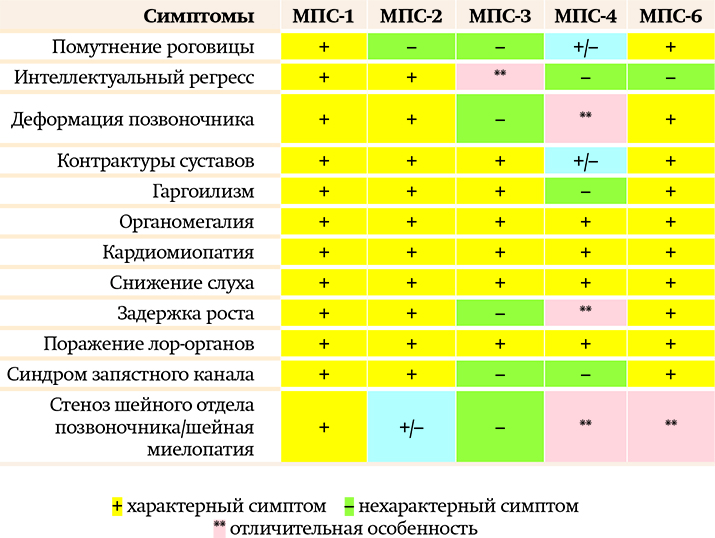
Таблица может помочь врачу на амбулаторном приеме предположить МПС, чтобы потом подтвердить/ опровергнуть диагноз лабораторными исследованиями
Шаг 2. Исключение других болезней накопления
Ряд болезней накопления имеет схожие с МПС внешние клинические признаки. Например, симптоматика альфа-маннозидоза включает:
- грубые черты лица;
- изменения скелета по типу множественного дизостоза;
- поясничный лордоз;
- диффузную мышечную гипотонию;
- шумное носовое дыхание;
- умеренную гепатоспленомегалию;
- нейросенсорную тугоухость;
- пупочную, паховую грыжи;
- умственную отсталость различной степени;
- частые респираторные инфекции, бронхиты, отиты.
Но для альфа-маннозидоза ведущим клиническим признаком является высокая степень нейросенсорной тугоухости, наличие признаков периферической полинейропатии (для МПС-1, МПС-2, МПС-6 характерен только синдром запястного канала), гуморальные и клеточные иммунодефициты. У пациентов с МПС, несмотря на то, что они склонны к частым респираторным инфекциям, иммунодефициты отсутствуют. Но главное отличие в результатах лабораторных исследований. При альфа-маннозидозе отмечается снижение активности альфа-маннозидазы в лейкоцитах. Накопление маннозосодержащих олигосахаридов. ГАГ в моче отсутствует. По результатам ДНК-диагностики заболевание обусловлено мутациями в гене MAN2B1.
Муколипидоз 2-го и 3-го типа также имеет симптомы, которые «роднят» его с разными типами МПС. Муколипидоз 2-го типа имеет более тяжелое течение и клинические проявления практически с первых месяцев жизни, быстрое прогрессирование. Муколипидоз 3-го типа мягче, отличается более поздним проявлением симптоматики и чуть меньшей скоростью прогрессирования. При этом вне зависимости от типа муколипидозу свойственны общие с мукополисахаридозом симптомы:
- грубый гаргоилизм;
- низкорослость;
- помутнение роговицы;
- тугоподвижность суставов;
- интеллектуальный дефицит;
- паховые и пупочные грыжи;
- органомегалия и др.
Отличить одно заболевание от другого помогает лабораторная диагностика. При муколипидозе 2-го типа отмечается повышение уровня лизосомных ферментов (N-ацетилгексозаминидазы) в плазме и низкий уровень в лейкоцитах крови. При муколипидозе 3-го типа — резкое снижение активности N-ацетилглюкозамин-1-фосфотрансферазы в лейкоцитах и культуре кожных фибробластов (ККФ). В отличие от МПС муколипидоз обусловлен мутациями в гене GNPTAB.
Фукозидоз — очень редкая патология, которая имеет общие с МПС симптомы: огрубление черт лица, низкорослость, органомегалию, грыжи, нейросенсорную тугоухость, контрактуры крупных и мелких суставов, помутнение роговицы и т. д.
Как и другие болезни накопления, фукозидоз может протекать с разной скоростью прогрессирования. При тяжелой форме болезни клинические проявления возникают уже на первом году жизни, быстро прогрессируют, приводя к тяжелой инвалидизации и раннему летальному исходу. При мягком течении клинические симптомы появляются в конце первого десятилетия, медленнее прогрессируют.
Главной отличительной особенностью фукозидоза являются специфические кожные проявления — ангиокератома, ангидроз. Наличие ангиокератомы вкупе с характерной для МПС симптоматикой дает основания предполагать фукозидоз. Окончательное слово за лабораторной диагностикой. При фукозидозе отмечается резкое снижение активности α-L-фукозидазы и ККФ, накопление гликолипидов и гликопротеинов в клетках. Молекулярно-генетическая диагностика указывает на мутации в гене FUCA1.
GM1-ганглиозидоз , в отличие от предыдущего заболевания, широко распространен среди болезней накопления. Несмотря на некоторые схожие с МПС симптомы (гаргоилизм, низкорослость, гиперплазия десен, костные деформации, органомегалия, тугоподвижность суставов и т. д.), GM1-ганглиозидоз имеет яркие отличительные особенности. В частности, ему свойственны макулодистрофия по типу «вишневой косточки», поражение почек, быстро прогрессирующее поражение ЦНС, эпилепсия, грубая задержка психомоторного развития, ангиокератома. По результатам лабораторной диагностики отмечается резкое снижение активности β-галактозидазы в лейкоцитах и ККФ. Заболевание обусловлено мутациями в гене GLB1. Болезнь практически всегда имеет тяжелое течение и неблагоприятный исход в первые годы жизни.
Множественная сульфатазная недостаточность — заболевание, которое легко спутать с МПС 2-го типа, если не выполнить объективную лабораторную диагностику. Клиническая симптоматика заболевания почти идентична симптоматике МПС-2, но результаты лабораторных исследований показывают принципиальные отличия. Так, при множественной сульфатазной недостаточности в крови снижен уровень не только идуронат-2-сульфатазы, но и других ферментов, в частности арилсульфатаз А, В, С. ДНК-диагностика показывает мутации в гене SUMF1.
Шаг 3. Исключение иных наследственных заболеваний или синдромальных состояний, схожих с болезнями накопления, в том числе МПС
Микроделеция Xq28, включающая ген IDS. Заболевание обусловлено делецией в хромосомной области, которая затрагивает ген IDS. Мутации этого гена лежат в основе МПС 2-го типа, поэтому неудивительно, что клинические признаки микроделеции Xq28 практически совпадают с симптомами МПС-2, включая высокий уровень ГАГ в моче. Примечательно, что ферментозаместительная терапия, назначаемая обычно при МПС-2, в этом случае также может давать положительную динамику. Но формально это все-таки не МПС-2, поскольку заболевание вызвано делецией, затрагивающей ген IDS, а не мутацией в нем.
Синдром Беквита — Видемана. Достаточно распространенный синдром, имеющий схожую с МПС симптоматику: интеллектуальный дефицит, органомегалию, грыжи. В то же время молекулярно-генетическое исследование показывает совершенно отличную от МПС картину: это синдромальное состояние, обусловленное изменениями в регионе 11р15, включающем гены факторов роста и гены-супрессоры опухолевого роста.
Х-сцепленная демиелинизирующая лейкодистрофия со спондилоэпифизарной дисплазией. Для этого заболевания характерны клинические признаки, схожие с симптоматикой болезней накопления: гаргоилизм, изменения скелета по типу множественного дизостоза (сколиоз, воронкообразная деформация грудной клетки), интеллектуальный дефицит, вальгусная деформация нижних конечностей, рецидивирующие отиты, тугоухость и другие. При этом уровень ГАГ в моче в пределах нормы, отсутствует дефицит лизосомных ферментов. Молекулярно-генетическое исследование указывает на специфическую мутацию в гене AIFM1, что и позволяет установить верный диагноз.
Шаг 4. Дифдиагностика с заболеваниями, имеющими схожую с МПС клиническую симптоматику
И, наконец, необходимо исключить заболевания, под которые маскируются МПС: ювенильный ревматоидный артрит; детский церебральный паралич; гипертензионно-гидроцефальный синдром; периферическая нейропатия; гепатит неясного генеза; гепатолиенальный синдром; врожденный порок сердца; хронический отит, аденоидит, тонзиллит; хронический обструктивный бронхит; муковисцидоз; бронхиальная астма; гипотиреоз; иммунодефицитное состояние; помутнение роговицы.
Надо отметить, что часть этих диагнозов в контексте МПС лишь симптомы, например, помутнение роговицы, порок сердца, аденоидит или отит. Гепатит неясного генеза — симптом органомегалии, увеличения печени; муковисцидоз, хронический обструктивный бронхит — проявления поражения нижних дыхательных путей у пациентов с МПС.
Поэтому за постановкой любого диагноза должен стоять тщательный сбор анамнеза, включая неонатальный и семейный, а также объективная лабораторная диагностика. В случае с МПС этот принцип исключительно важен. Вот лишь пару примеров. Раньше детям с МПС часто выставляли детский церебральный паралич как окончательный диагноз. И это на фоне отсутствия отягощенного неонатального анамнеза, хронической гипоксии, повышения мышечного тонуса, позотонических и патологических рефлексов, характерных для ДЦП.
К диагнозам, которые прежде часто «подменяли» истинный, относится и гипотиреоз. Отчасти заболевание действительно напоминает МПС (крупный язык, задержка психоречевого развития, увеличенная щитовидная железа по данным УЗИ могут навести на мысль о болезни накопления). Однако у пациентов с МПС увеличение щитовидной железы обусловлено отложением ГАГ в тканях органа, которое при этом не дает никакого гормонального дисбаланса.
К счастью, сегодня такие заблуждения скорее редкость.
Мукополисахаридозы
![]()

Мукополисахаридозы представляют собой один из видов лизосомальных болезней накопления Общие сведения о лизосомальных болезнях накопления Лизосомальные болезни накопления представляют собой редкие наследственные нарушения обмена веществ. Наследственные заболевания возникают, когда родители передают дефектные гены, вызывающие эти. Прочитайте дополнительные сведения , при которых сложные молекулы сахара не расщепляются, а накапливаются в опасном количестве. Результатом является характерный внешний вид и аномалии костей, глаз, печени и селезенки, иногда сопровождаемые умственной отсталостью. Мукополисахаридозы возникают, когда родители передают дефектные гены Гены Гены представляют собой сегменты дезоксирибонуклеиновой кислоты (ДНК) и содержат код для определенного белка, функционирующего в одном или нескольких типах клеток в организме. Хромосомы — это. Прочитайте дополнительные сведения , вызывающие эти нарушения, своим детям.
Мукополисахаридозы возникают, когда организму не хватает необходимых ферментов для расщепления и сохранения сложных молекул сахара.
Как правило, симптомы включают низкорослость, волосатость, тугоподвижность суставов пальцев и грубые черты лица.
Диагноз ставится на основании симптомов, результатов физикального обследования и анализов крови.
Хотя нормальная продолжительность жизни возможна, некоторые типы заболевания становятся причиной преждевременной смерти.
Лечение может включать пожизненную заместительную терапию ферментами, а иногда — трансплантацию костного мозга.
Сложные молекулы сахара, называемые мукополисахаридами (гликозаминогликанами), являются важнейшими составляющими многих тканей организма. При мукополисахаридозах организму не хватает необходимых ферментов для расщепления (преобразования) и сохранения мукополисахаридов. В результате избыток мукополисахаридов поступает в кровь и осаждается в аномальных местах по всему организму.
Типы мукополисахаридоза
В зависимости от того, какой фермент отсутствует, существует множество типов мукополисахаридоза. Типы обозначаются римскими цифрами и им иногда присваивают собственные имена (например, синдром Гурлер и синдром Хантера).
Симптомы мукополисахаридозов
При разных типах мукополисахаридоза отмечаются слегка отличающиеся симптомы, но, в целом, в младенчестве и детстве становятся заметными низкорослость, волосатость и аномалии развития. Лицо может казаться тяжелым, с большой головой, выпуклым лбом, коротким носом, а также большими губами и языком. Некоторые типы мукополисахаридозов вызывают умственную отсталость Умственная отсталость Ограничение умственных способностей — это серьезное снижение интеллектуальной деятельности ниже среднего показателя, присутствующее с рождения или раннего младенческого возраста и приводящее. Прочитайте дополнительные сведения , которая на протяжении жизни человека может усугубляться. При некоторых типах могут возникнуть нарушения зрения или слуха. Могут быть повреждены артерии или клапаны сердца. Суставы пальцев часто становятся тугоподвижными.
![]()
Диагностика мукополисахаридозов
Пренатальные скрининговые обследования
Диагноз ставится на основании симптомов и результатов обследования
рентгенологическое исследование костей;
Иногда визуализирующее обследование или другие анализы крови
После рождения врач обычно ставит диагноз мукополисахаридозов на основании симптомов и результатов физикального обследования. Наличие мукополисахаридоза у других членов семьи также говорит в пользу этого диагноза. При постановке диагноза могут помочь анализы мочи, но иногда они дают неточные результаты. Рентгенологическое исследование может показать характерные аномалии костей. Мукополисахаридозы также диагностируются с помощью анализа клеток крови. Имеется генетический анализ Генетический скрининг до беременности Генетический скрининг используется для того, чтобы определить, имеет ли пара повышенный риск рождения ребенка с наследственными генетическими отклонениями. Наследственные генетические расстройства. Прочитайте дополнительные сведения , позволяющий определить, имеет ли пара повышенный риск рождения ребенка с наследственным генетическим заболеванием.
Другие тесты проводятся в зависимости от симптомов. Например, дети, у которых имеются проблемы с сердцем, могут пройти визуализирующее обследование эхокардиографию Эхокардиография и другие процедуры с использованием ультразвука В ходе ультразвукового исследования используют высокочастотные (УЗИ) волны. Отраженные от внутренних структур волны регистрируются с получением движущегося изображения. Рентгеновские лучи в. Прочитайте дополнительные сведения  , а дети, у которых имеются проблемы со слухом, могут пройти аудиометрию Проведение анализов Во всем мире около полумиллиарда человек (почти 8 % населения мира) страдают потерей слуха. Более чем у 15 % жителей Соединенных Штатов Америки имеет место потеря слуха разной степени, влияющая. Прочитайте дополнительные сведения
, а дети, у которых имеются проблемы со слухом, могут пройти аудиометрию Проведение анализов Во всем мире около полумиллиарда человек (почти 8 % населения мира) страдают потерей слуха. Более чем у 15 % жителей Соединенных Штатов Америки имеет место потеря слуха разной степени, влияющая. Прочитайте дополнительные сведения  .
.
Прогноз при мукополисахаридозах
Прогноз зависит от типа мукополисахаридоза. Возможна нормальная продолжительность жизни. Некоторые типы, обычно те, которые затрагивают сердце, могут становиться причиной преждевременной смерти.
Лечение мукополисахаридозов
Заместительная терапия ферментами
В некоторых случаях трансплантация костного мозга или стволовых клеток
Для лечения некоторых мукополисахаридозов используется пожизненная заместительная терапия ферментами, которая может предотвратить усугубление заболеваний и привести к обратному развитию некоторых осложнений.
Дополнительная информация
Ниже приведены некоторые ресурсы на английском языке, которые могут быть полезными. Обратите внимание, что составители СПРАВОЧНИКА не несут ответственности за содержание этих ресурсов.
Национальная организация по редким заболеваниям (National Organization for Rare Disorders, NORD): этот ресурс предоставляет родителям и семьям информацию о редких заболеваниях, включая список редких заболеваний, группы поддержки и ресурсы клинических исследований.
Информационный центр по генетическим и редким заболеваниям (Genetic and Rare Diseases Information Center, GARD): этот ресурс предоставляет и информацию о редких или генетических заболеваниях в доступной для понимания форме.
ПРИМЕЧАНИЕ: Это — пользовательская версия ВРАЧИ: Просмотреть профессиональную версию
Просмотреть профессиональную версию
Авторское право © 2023 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Источник https://www.redkiebolezni.ru/mps/o-bolezni-mps/chto-takoe-bolezn-mps
Источник https://medvestnik.by/konspektvracha/differentsial-naya-diagnostika-mukopolisakharidozov
Источник https://www.msdmanuals.com/ru/%D0%B4%D0%BE%D0%BC%D0%B0/%D0%BF%D1%80%D0%BE%D0%B1%D0%BB%D0%B5%D0%BC%D1%8B-%D1%81%D0%BE-%D0%B7%D0%B4%D0%BE%D1%80%D0%BE%D0%B2%D1%8C%D0%B5%D0%BC-%D1%83-%D0%B4%D0%B5%D1%82%D0%B5%D0%B9/%D0%BD%D0%B0%D1%81%D0%BB%D0%B5%D0%B4%D1%81%D1%82%D0%B2%D0%B5%D0%BD%D0%BD%D1%8B%D0%B5-%D0%BD%D0%B0%D1%80%D1%83%D1%88%D0%B5%D0%BD%D0%B8%D1%8F-%D0%BE%D0%B1%D0%BC%D0%B5%D0%BD%D0%B0-%D0%B2%D0%B5%D1%89%D0%B5%D1%81%D1%82%D0%B2/%D0%BC%D1%83%D0%BA%D0%BE%D0%BF%D0%BE%D0%BB%D0%B8%D1%81%D0%B0%D1%85%D0%B0%D1%80%D0%B8%D0%B4%D0%BE%D0%B7%D1%8B