Синдром Гольденхара ( Окуло-аурикуло-вертебральная дисплазия )

Синдром Гольденхара — это редкое врожденное заболевание, которое проявляется множественными пороками развития, выраженным клиническим полиморфизмом. Возникает вследствие мутации генов, локализованных в 5, 14, 20 хромосомах. Для синдрома характерны разнообразные аномалии лицевого скелета, патологии органов чувств, зачастую болезнь сопровождается задержкой психического развития. План обследования включает генетическое тестирование для верификации диагноза, лабораторно-инструментальные методы с учетом ведущих клинических признаков. Лечение поддерживающее, многим больным требуется слухопротезирование, комплексная нейрореабилитация.
МКБ-10
Q87.0 Синдромы врожденных аномалий, влияющих преимущественно на внешний вид лица

- Причины
- Патогенез
- Симптомы
- Осложнения
- Диагностика
- Лечение синдрома Гольденхара
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Синдром назван в честь американского ученого Мориса Гольденхара, который описал его типичные клинические проявления в 1952 г. В 1963 г. Р.Дж. Горлин и его коллеги сообщили о собственных наблюдениях пациентов, дав болезни второе название «окуло-аурикуло-вертебральная дисплазия». Состояние выявляется с частотой от 1:3500 до 1:7000 живорожденных новорожденных, соотношение мальчиков и девочек составляет 3:2. Если в семье есть больной ребенок, то вероятность появления синдрома Гольденхара у следующих детей — не более 3%.
Синдром Гольденхара
Причины
Заболевание возникает при нарушении дифференцировки 1-2-й жаберных дуг, что провоцируется мутацией в генах GSC и TCOF1. В 98% случаев синдрома роль наследственности не удается проследить. Однако около 2% больных имеют родственников с аналогичной клинической симптоматикой. В литературе описаны случаи аутосомно-доминантного и аутосомно-рецессивного наследования.
Основными предрасполагающими факторами считаются:
- воздействие тератогенных факторов на ранних сроках гестации;
- наличие у матери сахарного диабета, избыточного веса;
- предшествующие искусственные прерывания беременности и выкидыши.
Патогенез
Механизмы формирования характерных для синдрома Гольденхара фенотипических изменений продолжают изучаться. На сегодняшний день ведущей является теория нарушений образований лицевых структур на ранних гестационных сроках (с 3-й по 8-ю неделю эмбриогенеза). Предположительно, в этот период в результате сочетанного воздействия неблагоприятных внешних факторов и генетических аномалий нарушается дифференцировка парных первой-второй жаберных дуг.
Как следствие, лобный, нижне- и верхнечелюстные эктодермальные отростки, происходящие из первой жаберной дуги, и ушные раковины, образованные 1 и 2 жаберными дугами, развиваются асимметрично. После рождения ребенка это проявляется специфическими изменениями верхней и нижней челюстей, глаз и глазниц, мимической и жевательной мускулатуры, структур наружного и среднего уха, аномалиями прикуса, дефицитом мягких тканей.
Симптомы
Основные признаки синдрома Гольденхара — аномалии строения лица, которые в 70% случаев являются правосторонними. У всех пациентов наблюдается асимметрия, недоразвитие нижней челюсти, уменьшение в размерах, деформация или отсутствие ушных раковин. Патологии органа слуха чаще бывают односторонними, сопровождаются атрезией слухового прохода, преаурикулярными выростами. Иногда формируется дополнительная рудиментарная ушная раковина.
Также характерно уменьшение размера глазных яблок (микрофтальмия), косоглазие, атрезия радужки, катаракта. Около 50% пациентов с болезнью Гольденхара имеют высокое готическое небо, широкий рот (макростомию), расщепление языка, аномальный прикус, отсутствие части зубов. В 40% случаев отмечаются косолапость, аномалии позвоночника (сколиоз, spina bifida), искривление ребер. У 30% больных возникают врожденные патологии внутренних органов: пороки сердца, гипоплазия легких, дисплазия почек.
Осложнения
Самым опасным последствием синдрома Гольденхара считается умственная отсталость, которая обусловлена двусторонней тугоухостью, снижением остроты зрения. При этом у большинства детей наблюдается нормальное функционирование нервной системы, однако сенсорная депривация создает затруднения при речевом и психомоторном развитии. Осложненное течение синдрома встречается при тяжелых врожденных пороках сердца, легких, почек.
Диагностика
В большинстве случаев предварительный диагноз устанавливается на основании характерных фенотипических признаков: деформации ушей, асимметрии лица, патологии развития нижней челюсти. Для подтверждения синдрома Гольденхара обязательно назначается консультация генетика, проводится комплекс диагностических мероприятий:
- Генетический анализ. Тестирование на специфическую мутацию генов 14q32, 5p15, MYT1 позволяет 100% подтвердить диагноз. Исследование выполняется в специальных генетических лабораториях с использованием методов секвенирования экзона, флуоресцентной гибридизации.
- Аудиометрия. Оценка слуха в динамике необходима всем пациентам, чтобы вовремя выявить кондуктивную тугоухость, обеспечить мероприятия по ее коррекции. Расширенное обследование у отоларинголога может включать импедансометрию, оценку рефлекса стапедиальной мышцы.
- Неврологический осмотр. Консультация детского невролога требуется для определения причин задержки психомоторного развития, оценки функционирования центральной и периферической нервной системы. При наличии показаний больного направляют на дообследование к психиатру.
- Инструментальная визуализация. Чтобы подтвердить или исключить соматические пороки, при синдроме Гольденхара проводятся рентгенография грудной клетки, эхокардиография, УЗИ органов брюшной полости. Для исследования ЦНС применяется КТ или МРТ головного мозга.
Лечение синдрома Гольденхара
Поскольку основной проблемой является тугоухость, в раннем возрасте рекомендовано слухопротезирование, чтобы предотвратить глухоту и возникающую на ее фоне задержку психоречевого развития. Также больной осматривается пластическим хирургом, чтобы по возможности скорректировать типичные внешние проявления синдрома, исправить асимметрию, гармонизировать черты лица.
Пациенты, у которых диагностирована болезнь Гольденхара, подлежат пожизненному динамическому наблюдению. Им назначаются регулярные обследования у офтальмолога, отоларинголога, невролога и других специалистов, чтобы контролировать состояние здоровья. Курсы поддерживающего лечения включают несколько направлений помощи, основными из которых являются:
- Метаболическая терапия. Для правильного развития ЦНС, улучшения когнитивных навыков используются ноотропы, витамины группы В.
- Нейрореабилитация. Чтобы улучшить речевое развитие, рекомендованы продолжительные курсы занятий с логопедами, сурдологами. По показаниям проводится психологическая коррекция.
- Специальное обучение. Пациентам с признаками ЗПР необходимо продолжать учебу в коррекционных классах с дефектологами, сурдопедагогами.
Прогноз и профилактика
При отсутствии у больного врожденных соматических пороков и достаточном уровне интеллекта прогноз благоприятный, большинство пациентов доживают до зрелого и пожилого возраста. Вызывают опасения случаи синдрома Гольденхара, которые сопровождаются кардиологическими, пульмонологическими или нефрологическими пороками, также неблагоприятный прогноз устанавливают при тяжелой умственной отсталости.
Основу профилактики синдрома составляет антенатальная охрана плода. Критическим периодом является первый триместр, поскольку именно в это время возникают характерные костные аномалии. Беременным женщинам рекомендовано избегать контакта с химикатами, рентгеновским излучением, а также соблюдать противоэпидемические меры для защиты от вирусных инфекций.
Литература
1. Клинический случай синдрома Гольденхара в психиатрической практике/ А.В. Ковалева// Acta Biomedica Scientifica. — 2020. — №3.
2. Клинический случай синдрома Гольденхара у новорожденного/ Л.Г. Киселева, Л.П. Мокеева, Ю.С. Тишкова, Н.В. Павловский// Вятский медицинский вестник. — 2015. — №46.
3. Диагностика синдрома Гольденхара в периоде новорожденности/ С.И. Мизинцева// Бюллетень Северного государственного медицинского университета. — 2012.
4. Особенности общеклинических проявлений синдрома Гольденхара/ И.А. Карякина// Системная интеграция в здравоохранении. — 2010. — №2.
Синдром Гольденхара — симптомы и лечение
Что такое синдром Гольденхара? Причины возникновения, диагностику и методы лечения разберем в статье доктора Гавран Надежды Александровны, генетика со стажем в 12 лет.
Над статьей доктора Гавран Надежды Александровны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина

Генетик Cтаж — 12 лет
Клиника «НоваВита»
Лаборатория «Геномед»
«Медико-генетический центр»
Дата публикации 27 июня 2020 Обновлено 26 апреля 2021
Определение болезни. Причины заболевания
Синдром Гольденхара — это редкая врождённая аномалия, при которой изменяются размеры и форма лицевых структур. Обычно изменения локализуются на одной стороне лица, вызывая его асимметрию, но иногда встречается двустороннее поражение.

Данный синдром относится к спектру врождённых аномалий черепа и лицевых структур, имеющих общий термин «краниофациальная микросомия». Под ним понимается уменьшение какой-либо структуры тела в пределах черепно-лицевой области.
Синонимы синдрома: окулоаурикулярная дисплазия, фацио-аурикуло-вертебральная ассоциация, синдром 1-й и 2-й жаберных дуг, отомандибулярный дизостоз, гемифациальная микросомия и др.
Приблизительная частота встречаемости синдрома Гольденхара — 1 случай на 3500-25000 новорождённых [9] . У мальчиков он встречается в 2 раза чаще, чем у девочек.
Точные причины заболевания на сегодняшний день до конца не известны [1] [2] [3] [4] . Большинство случаев возникают случайно в семьях без отягощённой истории болезни. Однако у 1-2 % пациентов с синдромом Гольденхара есть близкие родственники с подобным нарушением. Это свидетельствует о роли генетических факторов в возникновении данной патологии [4] [5] . В частности предполагается участие гена MYT1, расположенного в локусе q13.33 хромосомы 20.
Другим возможным фактором развития синдрома Гольденхара являются хромосомные аномалии — потеря или удвоение участка хромосомы. Как правило, у людей с этими нарушениями могут наблюдаются такие сочетанные пороки развития, как аномалии сердца, лёгких, почек, конечностей и центральной нервной системы [1] [2] [5] [6] .
Некоторые исследователи полагают, что формированию синдрома способствует нарушение кровотока или внешние повреждающие факторы:
- приём некоторых лекарственных препаратов, противопоказанных при беременности;
- вредные привычки;
- химические и физические агенты, воздействующие на плод на 3-8 неделе внутриутробного развития [5][6] .
Также нельзя исключить роль таких акушерско-гинекологических факторов, как предшествующие аборты, сахарный диабет и ожирение [18] .
Первые описания врождённых аномалий лицевых структур обнаружены в древних письменах, датированных 2000 лет до н. э. В Колумбии и Мексике были найдены древние керамические изделия с изображениями различных вариантов гемифациальной микросомии, в том числе наследственной: на одном из изделий был изображён родитель с ребёнком на руках, которые имели схожие аномалии лица [10] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы синдрома Гольденхара
Для синдрома Гольденхара характерна асимметрия лица (одностороннее недоразвитие челюсти) в сочетании с аномалиями ушных раковин, доброкачественными опухолями глаз и поражением спинного мозга (как правило в шейном отделе позвоночника). В большинстве случаев эти нарушения локализуются с правой стороны [19] . Однако до 30 % людей с синдромом Гольденхара имеют двусторонние аномалии лицевых структур.
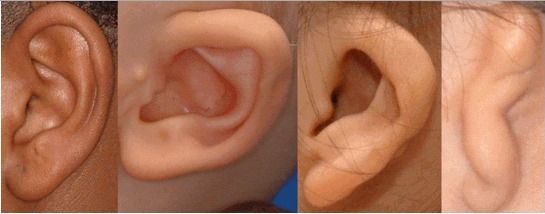
К лицевым аномалиям синдрома относятся:
- расщелины лица и нёба, аномалии лицевых мышц, верхней и нижней челюстей, скуловой и височной костей;
- аномалии ушных раковин: от недоразвития или полного отсутствия ушной раковины до образования околоушных кожных выростов при нормально сформированной ушной раковине;
- аномалии глаз (встречаются реже): одно- или двухстороннее уменьшение глазного яблока (микрофтальмия) вплоть до его отсутствия (анофтальмии), эпибульбарные дермоидные кисты глаз (доброкачественные опухоли) и ретинопатии [7] .
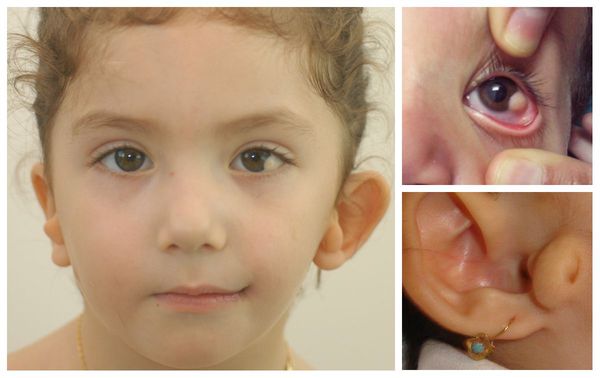
Перечисленные лицевые аномалии могут сопровождаться нарушением слуха, неправильной закладкой и прорезыванием зубов и другими нарушениями, которые могут повлиять на психофизическое развитие ребёнка.
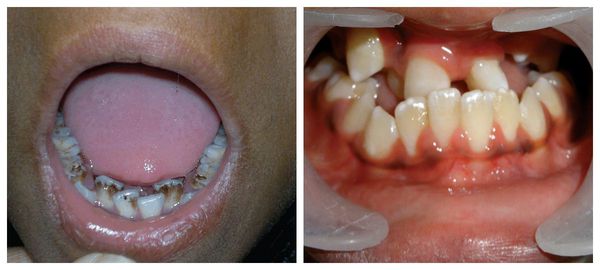
Патогенез синдрома Гольденхара
Лицевые структуры начинают формироваться на ранних сроках беременности. Со второй недели развития эмбриона на его головном конце образуется первичная ротовая ямка. К концу третьей недели она постепенно углубляется, достигает передней кишки (эндодермы) и, соединяясь с ней, образует начало пищеварительного тракта. В это же время по бокам головки эмбриона возникают два углубления — 1-я и 2-я жаберные щели, а ещё чуть позже — 3-я и 4-я щели. Между ними формируются жаберные или глоточные дуги, состоящие из нескольких частей: мешка, арки, бороздки и мембраны.
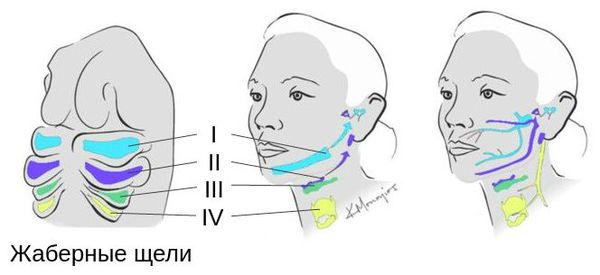
К концу первого месяца развития эмбриона первая жаберная дуга даёт начало пяти отросткам эктодермы: лобному, двум верхне- и нижнечелюстным. Непарный лобный отросток на третьей неделе разделяется на срединный и боковые носовые отростки, из которых к концу 10-11 недели внутриутробного развития формируются лоб, глазницы, нос, средние части верхней челюсти и верхней губы [11] [12] [14] . Нижнечелюстные отростки образуют единую структуру к концу четвёртой недели, а верхнечелюстные — на шестой неделе развития. Также на шестой неделе из парных латеральных закладок нижнечелюстной дуги формируется язык. На седьмой неделе верхнечелюстные отростки объединяются с лобными, в результате чего формируются губы.
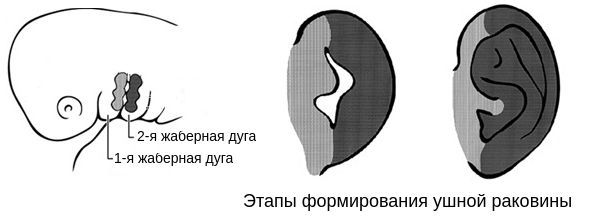
В образовании ушной раковины участвуют первая и вторая жаберные дуги. Из первой дуги образуется передняя треть наружного уха — козелок и ножки завитка. Срастание производных обеих дуг происходит очень рано: к восьмой неделе развития первичная ушная раковина оказывается уже сформированной, однако окончательный рельеф уха оформляется лишь к концу седьмого месяца развития эмбриона [13] .
Таким образом, верхняя и нижняя челюсти, жевательная и мимическая мускулатура, наружное ухо и костные структуры среднего уха формируются из первой и второй жаберных дуг с третьей по восьмую неделю развития эмбриона. Этот период является «критическим» в отношении возникновения пороков развития лица и челюстей. Нарушить нормальное развитие черепно-лицевых структур на данном этапе может сочетанное воздействие внешних факторов, хромосомных и генетических аномалий.
Классификация и стадии развития синдрома Гольденхара
Объём дефектов лицевых структур оценивается по классификации OMENS, в которой выделяют пять групп аномалий:
- O — поражение глазницы;
- M — недоразвитие нижней челюсти;
- E — аномалия уха;
- N — вовлечённость нерва;
- S — дефицит мягких тканей.
Степень тяжести данных дефектов определяется по классификации, созданной учёными Pruzansky S. и Kaban L. B.:
- 1 степень — уменьшение нижней челюсти и суставной ямки височной кости с сохранением анатомии других структур;
- 2а степень — деформация ветви нижней челюсти, суставного отростка и суставной ямки, сопровождается дефицитом жевательной мускулатуры, при этом функция височно-нижнечелюстного сустава сохраняется;
- 2б степень — недоразвитие и деформация мыщелка и суставной ямки, при этом височно-нижнечелюстной сустав не функционирует;
- 3 степень — отсутствие ветви нижней челюсти, мыщелка и суставной ямки с выраженным дефицитом мягких тканей на стороне поражения, височно-нижнечелюстной сустав не сформирован [16] .
Основываясь на своих многолетних наблюдениях, стоматолог-хирург Г. В. Кручинский выделил три варианта синдрома Гольденхара, каждый из которых подразделил на несколько типов:
- Синдром первой и второй жаберных дуг:
- односторонний ушной тип — лицо симметрично, наблюдаются аномалии ушной раковины;
- односторонний челюстно-лицевой и ушной тип (редко бывает двусторонним) — асимметрия лица из-за недоразвития челюстей и других прилегающих структур лёгкой и средней степени тяжести;
- односторонний черепно-челюстно-лицевой, суставной и ушной тип (редко бывает двусторонним) — выраженная асимметрия лица из-за тяжёлой степени недоразвития челюстей и прилегающий структур, отсутствия суставного отростка, головки и даже суставной ямки, атрофии подкожной клетчатки, слюнных желёз, мимических и жевательных мышц.
- Синдром первой жаберной дуги:
- односторонний нижнечелюстной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сохранением формы ушной раковины, сужением слухового прохода или свищом;
- односторонний или двусторонний нижнечелюстной и ушной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сужением слухового прохода и аномалией ушной раковины (её опущением, уменьшением и пр.).
- Простой синдром второй жаберной дуги:
- односторонний или двусторонний ушной тип — лицо симметрично, наблюдаются аномалии ушей в сочетании с дефектом мочек и лопоухостью.
По информации европейской базы данных редких заболеваний Orphanet [4] , все клинические проявления синдрома Гольденхара можно разделить на три группы:
- Очень частые (80-99 %):
- асимметрия лица;
- недоразвитие верхней челюсти;
- нарушение слуха;
- околоушные выросты (добавочные ушные раковины);
- уплощение лицевых скул.
- Частые (30-79 %):
- аномалии внутреннего и среднего уха;
- аномалии позвонков;
- аномалии ушных раковин (чаще односторонние), вплоть до недоразвития;
- атрезия (заращение) наружного слухового прохода;
- микрогнатия;
- нарушение грудного вскармливания;
- нарушение речи;
- расщелина нёба и/или верхней губы (заячья губа).
- Редкие (5-29 %):
- агенезия мозолистого тела (отсутствие проводящих путей между правым и левым полушариями);
- отсутствие одной или двух почек;
- аномалии гортани;
- аномалии рёбер;
- недоразвитие или отсутствие глаза, больших пальцев кистей;
- атрофия коры головного мозга;
- аутизм;
- вентрикуломегалия (увеличение мозговых желудочков);
- недоразвитие лёгких;
- аномалия расположения почек;
- недоразвитие части верхнего века (колобома);
- аномалия гортани и трахеи;
- макростомия (незаращение уголка рта);
- мышечная гипотония (слабость);
- нарушение зрения;
- низкий рост;
- пороки сердца (тетрада Фалло, дефект межжелудочковой перегородки);
- сколиоз;
- трахеопищеводный свищ;
- умственная отсталость.
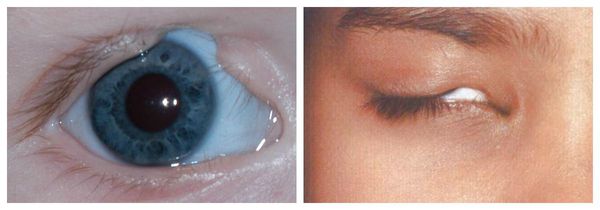
Осложнения синдрома Гольденхара
В раннем возрасте асимметрия нижней челюсти приводит к неправильному развитию и прогрессирующей деформации верхней челюсти и остальных структур лицевого скелета. Со временем ребёнку становится трудно жевать и глотать. При выраженном недоразвитии нижней челюсти у пациента могут возникнуть постоянные проблемы с дыханием, вплоть до апноэ во сне (остановки дыхания).
В целом расщелины лица и/или нёба, недоразвитие верхней и нижней челюсти, лицевых мышц, скуловой и/или височной костей способны вызывать проблемы с зубами, трудности при кормлении, нарушение речи и изменение эстетических параметров лица.
Аномалии ушных раковин в некоторых случаях сопровождаются атрезией (заращением) слухового канала либо полным его отсутствием, что приводит к нарушению слуха. Из-за этого ребёнку сложнее ориентироваться в пространстве, так как он не понимает, откуда исходит тот или иной звук.
Аномалии глаз, такие как дермоидные кисты глаз и колобомы (недоразвитие части верхнего века), способны приводить к нарушению зрительной функции вплоть до частичной или полной потери зрения [1] [4] [7] .
Диагностика синдрома Гольденхара
Как правило, диагностировать синдром Гольденхара не составляет труда. Постановка этого диагноза основана на оценке внешних признаков, клинической симптоматике и результатах дополнительных исследований — КТ, рентгенографии, МСКТ черепа, эхокардиографии и ультразвуковой диагностики. КТ, как правило, проводится для подготовки ребёнка к оперативному лечению.
Обязательной частью УЗИ плода на 18-20 неделе беременности является оценка лицевых структур. Методика исследования включает мультиплоскостную оценку положения и размеров орбит, структур глазных яблок, правильности формирования челюстей, носа и носогубного треугольника, расположения и формы ушных раковин. УЗИ лицевых структур плода позволяет выявить недоразвитие нижней челюсти, расщелины губы и/или нёба, отсутствие или недоразвитие глаза и ушной раковины [15] .
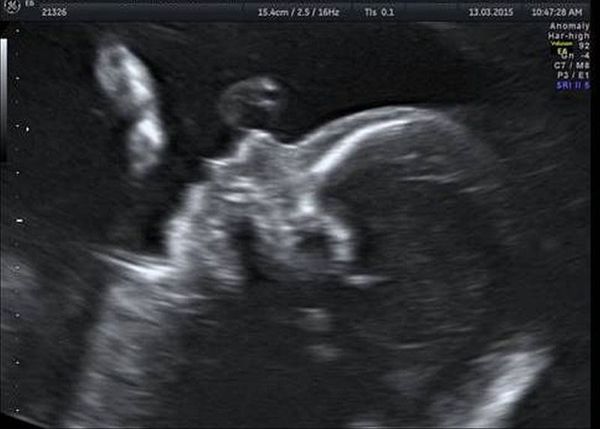
Генетическое тестирование может быть предложено для подтверждения диагноза, т. е. для исключения генетических состояний, включающих аналогичные лицевые аномалии, связанные с хромосомными и моногенными нарушениями. К таким заболеваниям относятся прогрессирующая гемиатрофия лица, синдром Нагера, челюстно-лицевой дизостоз и др. Однако минимальные диагностические критерии не установлены. Имеются описания единичных случаев диагностики данного синдрома с помощью тестирования до родов.
После рождения всем детям до наступления 6 месяцев во избежание задержки психоречевого развития проводится оценка слуха. Для этого выполняется измерение слуховых вызванных потенциалов: регистрация реакции мозга на звуковые раздражители. Зачастую на поражённой стороне у детей с синдромом Гольденхара выявляется тугоухость.
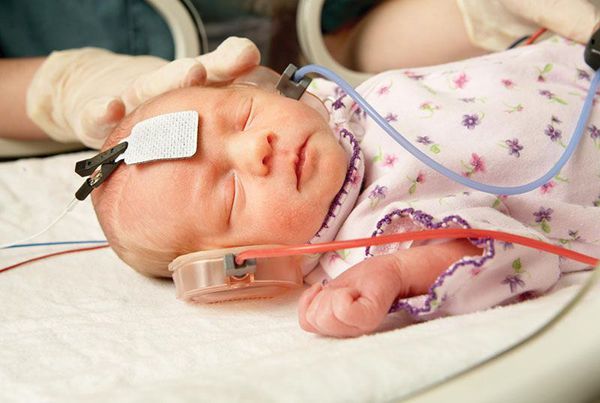
Лечение синдрома Гольденхара
Для лечения пациентов с синдромом Гольденхара применяются многоэтапные хирургические вмешательства, которые проводятся в разные периоды роста и развития черепно-лицевых структур. Лечение длительное, зависит от локализации и выраженности патологии. Оно направлено на восстановление формы и размеров челюстей, ушной раковины и других структур, а также на восстановление функций слуха, жевания и улучшение эстетических параметров лица [3] [6] [8] .
Лечение проявлений синдрома Гольденхара следует начинать как можно раньше. Своевременная коррекция челюстных нарушений у ортодонта способствует успешному хирургическому лечению в последующем и сохраняет баланс лицевого скелета.
Для устранения выраженных дефектов нижней челюсти применяют индивидуально-смоделированные эндопротезы либо костно-хрящевые аутотрансплантаты из рёбер, обладающие тенденцией к росту. Для устранения дефектов ушной раковины также используются силиконовые эндопротезы либо аутотрансплантаты.
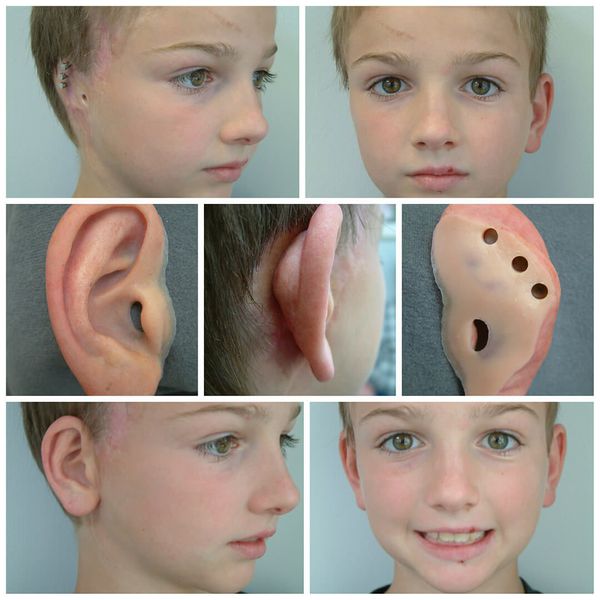
При выявлении нарушений слуха проводится слухопротезирование с помощью слуховых аппаратов либо альтернативными методами. Также необходимы регулярные занятия с сурдопедагогом и логопедом. Всё это позволяет предотвратить отставание ребёнка в речевом и общем развитии.
Решение проблем с кормлением заключается в применении специальных бутылочек и назогастрального зонда — трубки, которую вводят в желудок через нос.
Новообразования, локализующиеся на поверхности глазных яблок, могут быть удалены в случае нарушения зрения или при крупных размерах опухоли. У детей до 7 лет операция по удалению кисты проводится под наркозом. Врождённые пороки сердца, проблемы с почками и/или аномалии позвоночника также корректируются хирургическими методами [17] .
Прогноз. Профилактика
Прогноз жизни пациента с синдромом Гольденхара зависит от тяжести клинический проявлений, времени их диагностики и возможной коррекции. Долгосрочный прогноз предсказать сложно [13] .
Как правило, возникновение синдрома Гольденхара носит случайный, ненаследственный характер. При рождении больного ребёнка у здоровых родителей повторный генетический риск для потомства составляет не более 2-3 % [21] .
При отягощённом семейном анамнезе не исключён наследственный характер заболевания. В таком случае риск для потомства по краниофациальной микросомии повышен. Для оценки риска показано медико-генетическое консультирование. Однако отсутствие конкретного мутирующего гена, характерного для развития синдрома Гольденхара, не позволяет точно предсказать выраженность симптомов у потомства.
Первичная (массовая) профилактика синдрома Гольденхара, как и любой врождённой аномалии, заключается в информировании населения и полноценной дородовой подготовке, направленной на предупреждение возникновения заболевания.
Индивидуальная профилактика синдрома предполагает проведение медико-генетического консультирования семьи и пренатальной ультразвуковой диагностики беременной женщины в установленные сроки [12] .
Список литературы
- Hamosh A. Hemifacial Microsomia // Online Mendelian Inheritance in Man (OMIM). — 2019.
- Rollnick B. R., Kaye C. I. Oculo-auriculo-vertebral anomaly. On: Buyse M. D. Birth Defects Encyclopedia. Center for Birth Defects Information Services. Dover, MA: 1990; 1272-1274.
- Heike C. L., Luquetti D. V., Hing A. V. Craniofacial Microsomia Overview – Archived chapter, for historical reference only // GeneReviews. — 2009.ссылка
- Goldenhar syndrome // Orphanet. — 2014; 1-10.
- Tewfik T. L., et al. Manifestations of Craniofacial Syndromes // Medscape Reference. — 2019.
- Goldenhar Syndrome. FACES: The National Craniofacial Association. — 2016.
- Human Phenotype Ontology. Hemifacial hypoplasia. — 2018.
- National Organization for Rare Disorders (NORD). Oculo-Auriculo-Vertebral Spectrum. — 2007.
- Digilio M. C., et al. Congenital heart defects in patients with oculo-auriculo-vertebral spectrum (Goldenhar syndrome) // AJMG. — 2008; 146A (14): 1815-1819.ссылка
- Longacre J. J. Cranio-facial anomalies: Pathogenesis and Repair. — Philadelphia and Toronto, 1968.
- Беляков Ю. А. Стоматологические проявления наследственных болезней и синдромов. — М.: Медицина, 1993.
- Козлова С. И., Семанова Е., Демикова Н. С., Блинникова О. Е. Наследственные синдромы и медико-генетическое консультирование: справочник. — Л.: Медицина, 1989.
- Андреева О. В., Анохина А. В., Краснов М. В., Загребаева Е. А. и др. Медико-генетическое консультирование в стоматологии // Вестник Чувашского университета. — 2011. — № 3. — С. 262-268.
- Лазюк Г. И., Лурье И. В., Черствой Е. Д. Наследственные синдромы множественных пороков развития. — М.: Медицина, 1983.
- Гричанюк Д. А., Дубровская И. И., Лиштван Л. М. Эхографическое исследование как метод пренатальной и постнатальной диагностики при пороках развития челюстно-лицевой области // Медицинская панорама. — 2007. — № 2. — С. 63-67.
- Madrid J. R. P., Montealegre G., Gomez V. A New Classification Based on the Kaban’s Modification for Surgical Management of Craniofacial Microsomia // Craniomaxillofac Trauma Reconstr. — 2010; 3 (1): 1-7.ссылка
- Martelli-Junior H., de Miranda R. T., Fernandes C. M., Bonan P. R. F., et al. Goldenhar syndrome: clinical features with orofacial emphasis // J Appl Oral Sci. — 2010; 18 (6): 646-649.ссылка
- Карякина И. А. Особенности общеклинических проявлений синдрома Гольденхара // Системная интеграция в здравоохранении. — 2010. — № 2. — С. 18-31.
- Bielicka B., Necka A., Andrych M. Interdisciplinary treatment of patients with Goldenhar syndrome — clinical reports // Dent Med Probl. — 2006; 43: 458-462.
- Pinheiro A. L., Araujo L. C., Oliveira S. B., Sampaio M. C., Freitas A. C. Goldenhar’s syndrome — case report // Braz Dent J. — 2003; 14 (1): 67-70.ссылка
- Medina-de la Cruz A. Y., Rodríguez-Balderrama I., Burciaga-Flores C. H., de la O-Cavazos M. E. Spectrum of hemifacial microsomia in a pre-term newborn. Case presentation and literature review // Medicina Universitaria. — 2015; 17 (68): 158-161.
Синдром Гольденхара

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
- Код по МКБ-10
- Эпидемиология
- Причины
- Патогенез
- Симптомы
- Осложнения и последствия
- Диагностика
- Какие анализы необходимы?
- Дифференциальная диагностика
- Лечение
- К кому обратиться?
- Профилактика
- Прогноз

Окуло-аурикуло-вертебральная дисплазия, как еще называется эта достаточно редкая врожденная патология, обычно затрагивает развитие органов одной половины лица: глаз, уха, носа, мягкого неба, губ, челюсти. Она является отдельным видом лицевой микросомии, при которой из-за внутриутробного недоразвития скелетных, нервно-мышечных и прочих компонентов мягких тканей (производных I и II жаберных щелей) наружные органы одной половины лица отличаются заметно более мелкими размерами. Очень редко эта патология бывает двусторонней.

[1], [2], [3], [4], [5]
Код по МКБ-10
Q87.0 Синдромы врожденных аномалий, влияющие преимущественно на внешний вид лица
Эпидемиология
Медицинская статистика свидетельствует, что заболевания окуло-аурикуло-вертебрального спектра в структуре внутриутробных аномалий черепно-лицевой зоны следует за такими патологиями как заячья губа и волчья пасть, как сочетанных, так и отдельных. Частота появления синдрома Гольденхара, упоминаемая в зарубежной медицинской литературе, – один ребенок из 3,5-7 тысяч рожденных живыми младенцев. Этот синдром диагностируется у одного из тысячи глухих новорожденных. Примерно в 70% случаев поражения носят односторонний характер, при двусторонних дефектах они более выражены с одной стороны, причем среди них превалирует правая сторона с частотой встречаемости 3:2. Распределение по половому признаку – на троих детей мужского пола припадает две девочки.
[6], [7], [8], [9]
Причины синдрома Гольденхара
Сочетание дисплазии глаз, ушей и позвоночника, описанное в начале 50-х годов прошлого века американским доктором М.Гольденхаром, увековечилось его именем. Это редкая врожденная патология пока еще мало изучена, но мнение большинства исследователей совпадает в том, что этиологически он обусловлен генетической предрасположенностью. Тип наследования в настоящее время не определен, случаи заболевания носят спорадический характер. Есть сообщения об аутосомно-доминантном семейном наследовании. У пациентов с этим заболеванием выявляются хромосомные аномалии. Гипотетически факторы риска рождения ребенка с диспластическим поражениями окуло-аурикуло-вертебральных структур – это и кровнородственный брак, и предшествующие рождению ребенка аборты у матери, и тератогенез экзогенной или эндогенной природы, в частности, сахарный диабет или алиментарное ожирение будущей мамы.
Степень риска рождения больного ребенка у генетического носителя составляет 3%, а повторение рождения ребенка с этим дефектом в одной семье приравнивается к 1%.
[10], [11]
Патогенез
Патогенез, опять же, гипотетически, основан на возможности кровоизлияния на участке I и II жаберных щелей эмбриона, совпадающего по времени с заменой источника кровоснабжения в данной зоне. Подача крови из стременной артерии замещается снабжением из наружной сонной артерии. Сосудистый инсульт, произошедший в данное время и в данном месте, приводит к патологическим трансформациям клеточной пролиферации и аномальному формированию костно-мышечных, нервных и прочих элементов мягких тканей, развивающихся из производных I и II жаберных щелей.
[12], [13], [14], [15], [16], [17], [18], [19], [20]
Симптомы синдрома Гольденхара
Первые признаки наличия лицевой микросомии в основном определяются визуально при осмотре новорожденного. Типичными симптомами являются некоторая ассиметрия лица, нарушение размеров и положения глазницы, деформация ушных раковин в форме специфических аурикулярных «выступов», при этом другие изменения внешнего уха могут отсутствовать, недоразвитие нижней челюсти.
С ростом ребенка симптоматика становится все более заметной. Фенотип Голденхара включает аномалии развития уха (микротию), глаз, носа, мягкого неба, губ и челюсти. Одним из типичных симптомов считается наличие хористом (эпибульбарных дермоидов) на поверхности глазного яблока. Это опухолевые образования, содержащие не характерные для их локализации ткани (волосяные фолликулы, сальные и потовые железы, фиброжировую ткань). Данный симптом специфичен для 70% случаев синдрома Гольденхара. Из офтальмологических мальформаций могут присутствовать (25% случаев и более) – липодермоиды в наружной области конъюнктивы глазного яблока, коломбы верхнего века, пороки глазодвигательной мускулатуры, разрез глаз с опущенными вниз внешними уголками глазных яблок. Изредка (не чаще 5% случаев) наблюдается малый диаметр роговицы глаза, сквозной дефект или отсутствие радужной оболочки, опущение верхнего века, недоразвитие глазного яблока и его малые размеры, косоглазие и катаракта.
Аномалии развития ушных раковин встречаются чаще всего. Они деформированы и заметно меньше нормы по размеру (примерно 80% пациентов), у половины пациентов с синдромом имеют аномальное расположение, может отсутствовать наружный слуховой проход (40% пациентов). У 55 % больных наблюдались дефекты развития среднего уха и отсутствие слуха.
Очень характерный признак синдрома Гольденарха (85%) –недоразвитие отростков нижней челюсти, также достаточно часто асимметричны и недоразвиты лицевые мышцы, верхняя и нижняя челюсть. При осмотре ротовой полости наблюдается небо в форме высокой арки, иногда с расщелиной, открытый прикус, слишком широкая ротовая щель, расщелина язычка и добавочные уздечки.
Чуть меньше половине случаев сопутствовали недоразвития позвонков, чаще шейного отдела – клиновидные, слитые, полупозвонки, сколиоз, трети – spina bifida, пороки развития ребер, пятой части – косолапость.
Менее трети случаев синдрома Гольденхара сопровождались аномалиями развития сердечно-сосудистой системы (пороком межжелудочковой перегородки, открытым Боталловым протоком, тетрадой Фалло, сужением либо полным перекрытием аорты). Умственная отсталость в разной степени наблюдалась у десятой части пациентов с этим синдромом.
Существует несколько классификаций, отражающих стадии заболевания, а точнее степени его тяжести. Наиболее полная – OMENS. Она выделяет три стадии выраженности поражения каждого из объектов мальформаций при гемифациальной микросомии: глаза (orbit), нижняя челюсть (mandible), ухо (ear), лицевой нерв (facial nerve) и кости скелета (skeletal). Поскольку дефекты множественные и каждая структура обычно поражена в разной степени, то выглядит это примерно так: O2M3E3N2S1*. Звездочка отражает наличие дополнительных пороков нечерепно-лицевых объектов.
Классификация SAT обращает внимание на три основных объекта: скелет (skeletal), раковина уха (auricle), мягкие ткани (soft tissue). Согласно данной классификации пороки развития скелета рассматриваются по пяти стадиям (от S1 до S5), нарушения структуры ушной раковины – по четырем (от АО до A3); дефекты мягких тканей – по трем (от Т1 до Т3). Так, самая легкая стадия заболевания – S1A0T1, тяжелые пороки развития – S5A3T3. Система SAT проигрывает в сравнении с предыдущей по отсутствию важных объектов поражения, которые в ней не отражаются.
Некоторые авторы выделяют виды гемифациальной микросомии по фенотипу, связанному с объектами поражений. В этой классификации тип Гольденхара выделен в отдельный вид с присущими ему специфическими пороками развития.
[21], [22], [23], [24]
Осложнения и последствия
Последствия и осложнения этой врожденной патологии находятся в прямой зависимости от степени тяжести пороков развития, некоторые из которых несовместимы с жизнью, другие же, например, неправильный прикус – могут вызвать ряд неудобств. Очень много зависит от своевременно начатого лечения. Если время упущено, то у ребенка с данной патологией проявляется гипоплазия лицевых костей, причем прогрессирующая и все более заметная. Становится трудно совершать глотательные и жевательные движения. Патологии зрения, слуха тоже будут прогрессировать. Итогом всех ухудшений будут серьезные физические неудобства и психологический дискомфорт, что скажется на качестве жизни ребенка и его родителей.
[25], [26], [27], [28], [29], [30]
Диагностика синдрома Гольденхара
Как правило, предварительный диагноз этой врожденной аномалии устанавливается уже у новорожденного при выявлении асимметрии лица в сочетании с другими специфическими визуальными симптомами.
Для уточнения диагноза данного заболевания используются разнообразные диагностические процедуры. Одна из первых – определение остроты слуха, поскольку поражения наружного и внутреннего уха встречаются почти в каждом случае и в первую очередь обращают на себя внимание. Раннее исследование слуха вызвано необходимостью профилактики отставания ребенка в психоречевом развитии. В раннем возрасте ребенка диагностируют во время сна. Используются такие методы: импедансометрия, регистрация слуховых вызванных потенциалов (электрокохлеография, отоакустическая эмиссия), компьютерная аудиометрия.
Детей постарше тестируют в игровой форме с помощью речевой аудиометрии. Инструментальную и субьективную диагностику слуха показано проводить каждые шесть месяцев на протяжении семи лет.
Первоочередные консультации должны быть у челюстно-лицевого хирурга, офтальмолога, ортодонта, ортопеда, отоларинголога, чтобы диагностировать максимально возможные пороки развития. Инструментальную диагностику и анализы специалисты назначают по необходимости, в зависимости от выявленных патологий. Обычно назначают электрокардиографию, ренгенографию, ультразвуковое исследование внутренних органов.
Ребенку после достижения трехлетнего возраста назначается компьютерная томография височных зон.
Дети, имеющие этот диагноз, требуют консультаций многих специалистов в зависимости от наличия пороков развития: сурдолога, логопеда-дефектолога, кардиолога, нефролога, невропатолога и прочих.
[31], [32], [33]
Какие анализы необходимы?
Дифференциальная диагностика
Дифференциальная диагностика проводится с другими врожденными черепно-лицевыми пороками развития, такими как: дизостозы – нижнечелюстно-лицевой, гемифациальный, акрофациальный, другие виды гемифациальной микросомии, синдромы – Кауфмана и рото-лице-пальцевой, ассоциация Чарджа.
К кому обратиться?
Лечение синдрома Гольденхара
Многообразие пороков развития черепа и позвоночника, а также других органов и систем у пациентов с данной врожденной патологией приводит к многоэтапному лечению у многих специалистов. При легких степенях поражения ребенка наблюдают до трех лет, а затем начинается хирургическое лечение.
В случаях тяжелых врожденных дефектов сначала применяется оперативное лечение (в младенчестве или до достижения двух лет). После этого проводят симптоматическое комплексное лечение. Синдром Гольденхара без многоэтапного хирургического вмешательства вылечить нельзя. Количество и объем хирургических операций зависят от степени тяжести патологий. Таким больным проводят обычно компрессионно-дистракционный остеосинтез; эндопротезирование височно-нижнечелюстного сустава, нижней и верхней челюсти; остеотомию носа, нижней и верхней челюстей, исправляющую пороки их развития и патологический прикус; пластические операции (гениопластика, ринопластика). Для профилактики воспалительных осложнений и ускорения процесса реабилитации назначается антибиотикотерапия и витаминотерапия. При челюстно-лицевых хирургических манипуляциях назначаются, как правило, остеотропные антибиотики: пенициллины, линкомицин, эритромицин.
Пенициллины – естественные соединения, синтезируемые разными формами пенициллинового плесневого гриба и полусинтетические, на основе 6-аминопенициллановой кислоты, выделенной из естественных соединений. Их антибактериальная способность основана на нарушении клеточной оболочки бациллы. Они малотоксичны, имеют широкий спектр дозировок, однако, большинство лекарственных аллергий вызвано именно антибиотиками пенициллинового ряда.
Линкомицин – антибиотик выбора при аллергии к пенициллинам, может назначаться детям с месячного возраста, терапевтические дозировки препарата оказывают бактериостатический эффект, более высокие – бактерицидный, применяется для устранения инфицирования костей, суставов, мягких тканей. Противопоказан при тяжелых нарушениях почечной и печеночной функции. Может вызывать аллергию.
Эритромицин – относится к группе макролидных анибактериальных средств, имеет широкий спектр бактерицидного действия, может применяться в офтальмологии. Детям назначается с года. Одно из побочных действий этого препарата, а также реакция на передозировку – нарушение слуха, однако, оно считается обратимым. Поэтому препарат может быть назначен при непереносимости двух предыдущих, тем более, что профилактическая антибиотикотерапия назначается кратковременно. Ее цель – достичь к моменту операции наибольшей терапевтической плотности препарата в тканях. Профилактическое лечение начинают за час или два перед операцией и прекращают через двое-трое суток.
В зависимости от наличия болевых ощущений назначаются анальгетики. Маленьким пациентам назначают детский Нурофен, обладающий быстродействием, а также обеспечивающий жаропонижающий и противовоспалительный эффект и достаточно продолжительное действие (до восьми часов). Максимальная дозировка не должна превышать 30мг на килограмм веса ребенка ежесуточно.
В реабилитационный период нужно обеспечить ребенку полноценное питание и витамины. Назначаются поливитаминные комплексы, включающие аскорбиновую кислоту, ретинол, токоферол, витамины группы D и B.
Для предупреждения инфицирования и рассасывания послеоперационных отеков и инфильтратов применяют физиотерапевтическое лечение ультрафиолетовым излучением, ультразвуковыми и электромагнитными волнами, а также – лазерную и магнитную терапию и их сочетание, гипербарическую оксигенацию.
Ортодонтическое лечение предусматривают профилактику ассиметричного развития челюстей, исправление аномального прикуса, предоперационную подготовку зубов и мимической мускулатуры к операциям. Лечение у ортодонта делится на этапы, соответствующие трем видам прикуса:
- молочному – самый ответственный этап лечения, поскольку первый; ребенка и его родителей знакомят с патологией, вероятностью осложнений и правилами ухода за полостью рта, аппаратами для исправления челюстных дефектов, происходит привыкание к необходимым процедурам:
- сменному – на этом этапе основной задачей является исправление прикуса, профилактика и корректировка пороков развития челюстей;
- постоянному – на этом этапе продолжаются начатые мероприятия, съемные аппараты заменяются на брекет-системы, различные фиксаторы в зависимости от необходимости.
Ретенционные мероприятия, завершающие лечение, проводятся для закрепления достигнутого и заканчиваются в 18 лет, когда тело уже практически сформировано. Все это время до достижения совершеннолетия ребенок находится под контролем врачей, в зависимости от степени тяжести заболевания по необходимости ему назначаются лекарства, витамины и разнообразные процедуры. В комплекс лечения обычно входит лечебная гимнастика и работа с сурдопедагогом и психологом.
Альтернативная медицина
Пороки развития черепных и позвоночных структур при синдроме Гольденхара предполагают хирургическое вмешательство, однако, народное лечение может стать дополнительным хорошим подспорьем в реабилитационный период. Хочется только напомнить, что возможность использования народного метода необходимо сначала обговорить с лечащим врачом.
Лечебная гимнастика для исправления прикуса
Эти упражнения нужно делать в течение дня не менее двух раз, каждое из них повторить не менее шести раз:
- открыть рот как можно шире, посчитать в этом положении до десяти и резко его закрыть;
- исходное положение: прикоснувшись кончиком языка к небу, отвести его, не отрывая от неба, максимально вглубь – теперь несколько раз как можно шире открывать рот и закрывать его;
- сесть, поставить локти на стол, плотно упереть подбородок в сложенные горизонтально друг на дружку ладони – несколько раз открыть и закрыть рот (нижняя челюсть должна неподвижно находиться на ладонях).
Полезно также нагружать челюсти, регулярно хорошо пережевывая твердую пищу.
Дермоидные кисты, характерные для синдрома Гольденарха, лечатся только хирургическим путем.
Однако есть и народные методы избавления от кист. Рекомендуется очищать глаза отварами лекарственных трав: например, сделав отвар из цветков василька, листьев подорожника и семян тмина, закапывать по три капли в каждый глаз не менее пяти раз за день.
Можно промывать глаза чайной заваркой или отваром цветков ромашки аптечной: на стакан кипятка – три столовых ложки цветков.
Другой вполне безопасный и витаминный рецепт: смешать 1:1 мед и сок из ягод калины. Первую неделю принимать по одному грамму смеси натощак по утрам (в чайной ложке без верха – восемь граммов меда), на второй неделе – увеличить дозу вдвое, на третьей – еще вдвое, на четвертой – дозировка составляет 10г меда. Затем делают перерыв и повторяют прием в обратной последовательности, начиная с 10г.
При тугоухости тоже применяют лечение травами:
- две чайные ложки корня аира залить кипятком в объеме 600мл, настоять 2-3 часа, пить по три столовые ложки перед тремя приемами пищи в течение месяца, можно повторить спустя две недели;
- ½ стакана семян аниса долить до верха маслом из плодов шиповника, оставить в темном прохладном месте на три недели; затем процедить и закапывать на протяжении месяца, можно повторить спустя две недели;
- заваривать и пить без ограничений чай из лепестков роз, он тонизирует стенки сосудов и стимулирует микроциркуляцию крови в ушах.
Гомеопатия не сможет заменить оперативные вмешательства, необходимые при множественных пороках развития, присущих синдрому Гольденарха, однако, гомеопатические препараты могут поспособствовать быстрому восстановлению после операции (Арсеникум альбум, Стафизагрия). Отдельные отклонения от нормы, такие как тугоухость (Астэриас рубенс), косоглазие (Танацэтум), новообразования на голове, веках (Крокус сативус, Графитэс, Туя), а также общее состояние больного вполне могут быть сорректированы, особенно после консультации у врача-гомеопата.
[34], [35], [36], [37], [38]
Источник https://www.krasotaimedicina.ru/diseases/genetic/Goldenhar-syndrome
Источник https://probolezny.ru/sindrom-goldenara/
Источник https://ilive.com.ua/health/sindrom-goldenhara_123324i88403.html